Interferones beta versus acetato de glatirámero para la esclerosis múltiple recurrente remitente
Resumen
Antecedentes
Los interferones‐beta (IFN) y el acetato de glatirámero (AG) fueron los dos primeros tratamientos modificadores de la enfermedad (TME) aprobados hace 20 años para el tratamiento de la esclerosis múltiple (EM). Las tasas de prescripción de TME como primer tratamiento o como tratamiento de cambio y sus costos han aumentado de manera significativa durante la última década. Debido al aumento de la disponibilidad de TME, la elección de un TME específico debe reflejar el perfil de riesgos/beneficios, así como el impacto en la calidad de vida. Debido a que los grupos de pacientes con EM incluidos en diferentes estudios pueden variar de forma significativa, los ensayos de comparación directa se consideran el mejor enfoque para obtener datos objetivos fidedignos al comparar dos fármacos diferentes. La finalidad de esta revisión sistemática es resumir la evidencia disponible sobre la efectividad comparativa de los IFN beta y el AG en el curso de la enfermedad a través del análisis de los ensayos de comparación directa.
Ésta es una actualización de la revisión Cochrane "Interferones beta versus acetato de glatirámero para la esclerosis múltiple recurrente remitente" (publicada por primera vez en The Cochrane Library 2014, número 7).
Objetivos
Evaluar si los IFN‐beta y el AG difieren en cuanto a la seguridad y la eficacia en el tratamiento de los pacientes con EM recurrente remitente (EMRR).
Métodos de búsqueda
Se hicieron búsquedas en el registro del Grupo Cochrane de Esclerosis Múltiple y Enfermedades Raras del Sistema Nervioso Central (Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group) (8 agosto 2016) y en las listas de referencias de los artículos recuperados. Se estableció contacto con los autores y las compañías farmacéuticas.
Criterios de selección
Ensayos controlados aleatorizados (ECA) que compararan directamente los IFN beta versus AG en participantes en estudio afectados por EMRR.
Obtención y análisis de los datos
Se utilizaron los procedimientos metodológicos estándar previstos por Cochrane.
Resultados principales
Se incluyeron seis ensayos y cinco ensayos contribuyeron a esta revisión. Se asignó al azar a un total de 2904 participantes a los IFN (1704) y al AG (1200). La duración del tratamiento fue de tres años en un estudio, de dos años en los otros cuatro ECA, mientras que un estudio se detuvo de manera precoz (tras un año). Los IFN analizados en comparación con AG fueron IFN beta 1b 250 mcg (dos ensayos, 933 participantes), IFN beta 1a 44 mcg (tres ensayos, 466 participantes) e IFN beta 1a 30 mcg (dos ensayos, 305 participantes). Los participantes incluidos estaban afectados por EMRR activa. Todos los estudios presentaron un alto riesgo de desgaste. Tres ensayos siguen en curso, uno de ellos está completado.
Ambos tratamientos mostraron una eficacia clínica similar a los 24 meses, al tener en cuenta las variables de resultado primarias (número de participantes con recurrencias [riesgo relativo (RR) 1,04; intervalo de confianza (IC) del 95%: 0,87 a 1,24] o la progresión [RR 1,11; IC del 95%: 0,91 a 1,35]). Sin embargo, la evidencia de un único estudio indican que a los 36 meses las tasas de recurrencia fueron mayores en el grupo que recibió IFN que en el grupo de AG (RR 1,40; IC del 95%: 1,13 a 1,74; valor de p 0,002).
El análisis secundario de los resultados de la imaginología de resonancia magnética (IRM) mostró que los efectos sobre las lesiones nuevas o agrandadas T2 o de T1 resaltadas con contraste a los 24 meses fueron similares (diferencia de medias [DM] ‐0,15; IC del 95%: ‐0,68 a 0,39; y DM ‐0,14; IC del 95%: ‐0,30 a 0,02; respectivamente). Sin embargo, la reducción del volumen de la lesión ponderado en T2 o T1 fue significativamente mayor en los grupos que recibieron IFN que en los grupos de AG (DM ‐0,58; IC del 95%: ‐0,99 a ‐0,18; valor de p 0,004; y DM ‐0,20; IC del 95%: ‐0,33 a ‐0,07; valor de p 0,003; respectivamente).
El número de participantes que abandonaron el estudio debido a los eventos adversos fue similar en los dos grupos (RR 0,95; IC del 95%: 0,64 a 1,40).
La calidad de la evidencia para los resultados primarios se consideró moderada para las variables principales de evaluación clínicas, aunque para la seguridad y algunos resultados de la IRM (número de lesiones T2 activas), la calidad se consideró baja.
Conclusiones de los autores
Los efectos del IFN beta y el AG en el tratamiento de los pacientes con EMRR, incluidas las medidas clínicas (p.ej. pacientes con recurrencia, riesgo de progresión) y de la IRM (lesiones resaltadas con Gd), parecen ser similares o mostrar sólo diferencias pequeñas. Al considerar la acumulación de carga de la lesión en la IRM, el efecto de los dos tratamientos difiere en cuanto a que se encontró que los IFN beta limitan el aumento de la carga de la lesión en comparación con el AG. No hubo evidencia suficiente para realizar una comparación de los efectos de los dos tratamientos sobre los resultados informados por los pacientes, como las medidas de la calidad de vida.
PICOs
Resumen en términos sencillos
Una comparación de la efectividad y seguridad de los dos tratamientos más frecuentes para los pacientes con esclerosis múltiple (EM)
Antecedentes
Ésta es una actualización de la revisión Cochrane "Interferones beta versus acetato de glatirámero para la esclerosis múltiple recurrente remitente" (publicada por primera vez en The Cochrane Library 2014, número 7).
Actualmente la elección terapéutica para personas con EM requiere discusiones infromadas sobre la equivalencia de los agentes terapéuticos. Este estudio es importante debido a que es el primer informe que evalúa mediante comparación directa si dos tratamientos, interferones‐beta (IFN‐beta) y acetato de glatirámero (AG), presentan el mismo efecto sobre el curso de la enfermedad.
Características de los estudios
Se efectuaron búsquedas en las bases de datos médicas para obtener estudios en los que ni los participantes ni los investigadores conocieran qué tratamiento se estaba administrando (ensayos aleatorizados doble ciego). La eficacia de los dos tratamientos se consideró en cuanto a la aparición de recurrencias y la progresión de la enfermedad.
Resultados clave y calidad de la evidencia
Hasta agosto de 2016 se encontraron seis estudios con 2904 participantes (1704 tratados con IFN; 1200 con AG) que reunieron los requisitos de los criterios de inclusión. Se encontró que los dos tratamientos parecían tener efectos similares o sólo diferencias pequeñas en cuanto a la aparición de recurrencias o la progresión.
En términos generales, la calidad de la evidencia fue moderada, aunque en cuanto al perfil de seguridad, la calidad de la evidencia fue baja. Se encontró que el riesgo de datos de resultado incompletos fue alto, debido a que algunos estudios presentan un informe incompleto de los eventos adversos y a los números de participantes que abandonaron el estudio.
Cabe destacar que todos los estudios excepto uno fueron patrocinados por la industria farmacéutica. Además, todos los estudios se realizaron a corto plazo, la duración del tratamiento fue de tres años en un estudio y de dos años en los otros cuatro, mientras que un estudio se detuvo de manera precoz al año.
Authors' conclusions
Summary of findings
| interferons compared with glatiramer acetate for participants with relapsing‐remitting multiple sclerosis | ||||||
| Patient or population: people with relapsing‐remitting multiple sclerosis | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk (control) | Corresponding risk (intervention) | |||||
| Glatiramer acetate | Interferons | |||||
| Number of participants with relapse (M‐H, random, 95% CI) | Study population | RR 1.04 | 2184 | ⊕⊕⊕⊝ | Detection bias risk for clinical outcomes was judged as high for 1 study and low for the other 2 RCTs | |
| 36 per 100 | 38 per 100 | |||||
| Moderate | ||||||
| 35 per 100 | 36 per 100 | |||||
| Number of participants with confirmed progression (M‐H, random, 95% CI) | Study population | RR 1.11 | 2169 | ⊕⊕⊕⊝ | Detection bias risk for clinical outcomes was judged as high for 1 study and low for the other 2 RCTs | |
| 15 per 100 | 16 per 100 | |||||
| Moderate | ||||||
| 15 per 100 | 17 per 100 | |||||
| Number of participants who dropped out for AEs (M‐H, random, 95% CI) | Study population | RR 0.95 | 2685 | ⊕⊕⊝⊝ | ||
| 4 per 100 | 4 per 100 | |||||
| Moderate | ||||||
| 5 per 100 | 5 per 100 | |||||
| Mean number of active T2 lesions | 0.15 lower in IFN versus GA groups | 1790 | ⊕⊕⊝⊝ | Detection bias risk for MRI outcomes was judged as low for all studies | ||
| Mean number of new enhancing lesions | 0.14 lower in IFN versus GA groups | 1734 | ⊕⊕⊕⊝ | Detection bias risk for MRI outcomes was judged as low for all studies | ||
| Mean change in total T2‐hyperintense lesion load | 0.58 lower in IFN versus GA groups | 1608 | ⊕⊕⊕⊝ | Detection bias risk for MRI outcomes was judged as low for both studies | ||
| Mean change in total T1‐hypointense lesion load | −0.20 lower in IFN versus GA groups (−0.33 to −0.07) | 1602 | ⊕⊕⊕⊝ | Detection bias risk for MRI outcomes was judged as low for both studies | ||
| *The basis for the assumed risk (e.g. median control group risk (GA) across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group (IFNs) and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aHigh risk of attrition bias. | ||||||
Background
This is an update of the Cochrane review 'Interferons‐beta versus glatiramer acetate for relapsing‐remitting multiple sclerosis' (first published in The Cochrane Library 2014, Issue 7).
Description of the condition
Multiple sclerosis (MS) is the most important non‐traumatic cause of neurological disability in young adults. It is widely considered an immune‐mediated inflammatory disease of the central nervous system (CNS). The overall incidence rate of MS is between 3.5 and 6.6 per 100,000, and the prevalence is between 100 and 120 people per 100,000 (Alonso 2008; Richards 2002). After the first clinical episode (clinically isolated syndrome (CIS)), about 50% of people develop MS within six years (Chard 2011). MS commonly follows a relapsing‐remitting (RR) course, but after 10 years, about 50% of people with RRMS convert to a secondary progressive (SP) phase (Weinshenker 1989), which is characterised by progressive disability with or without superimposed relapses.
Disease‐modifying therapies (DMTs) are approved worldwide as therapeutic options for RRMS (Goodin 2008b; Weinstock‐Guttman 2008), although recent systematic reviews of placebo‐controlled trials have questioned their effectiveness, showing partial efficacy for exacerbation and disease progression (La Mantia 2010; Rice 2001). The first two DMTs, recombinant interferons‐beta (IFNs‐beta) and glatiramer acetate (GA), became available 20 years ago. In the Canadian provinces between 2002 and 2007, subcutaneous IFN‐beta 1a was the most commonly used drug, whereas GA showed the greatest growth in use (Rotstein 2010). Interferon‐beta 1a gained the greatest number of users in the United States between 2002 and 2008, whereas GA showed the greatest growth in use for therapy switches (Oleen‐Burkey 2013).
Today these drugs remain in worldwide use for the treatment of MS (Jelinek 2015). Persistence with injectable DMTs was found to be high in an 18‐year population‐based study, median time to discontinuation being 11.1 years, with a stop/switch rate of 46% for GA and 56% for IFN‐beta (Zhornitsky 2015).
Description of the intervention
Recombinant IFN‐beta products licensed for MS treatment include two IFN‐beta 1b preparations (Betaferon® (Bayer) (FDA 1993); and Extavia® (Novartis) (FDA 2009)) and three IFN‐beta 1a preparations: Avonex® (Biogen Idec) (FDA 1996); Rebif® (EMD Serono/Pfizer) (FDA 2002); and Plegridy® (Biogen Idec) (FDA 2014).
IFN‐beta 1b is a lyophilised protein produced by DNA recombinant technology using Escherichia coli. The approved dose is 250 mcg every other day by subcutaneous (SC) injection.
IFN‐beta 1a is a lyophilised glycoprotein produced in mammalian cells by using the natural human gene sequence. Three preparations are licensed for the treatment of MS: 30 mcg once a week administered by intramuscular (IM) injection (Avonex®); 22 or 44 mcg administered three times a week by SC injection (Rebif®); and 125 micrograms injected subcutaneously every 14 days (peginterferon‐beta 1a, Plegridy®)
GA, formerly known as co‐polymer‐1, is a random polymer of four amino acids: L‐alanine, L‐glutamic acid, L‐lysine and L‐tyrosine. It was the first non‐IFN drug approved for the treatment of MS (FDA 2001). The approved dose of GA (Copaxone® (Teva Pharmaceuticals)) is 20 mg administered subcutaneously every day.
How the intervention might work
Large placebo‐controlled, randomised double‐blind trials have demonstrated the efficacy of different forms of IFN‐beta in study participants with RRMS, as assessed by clinical evaluation and magnetic resonance imaging (MRI) (IFN MSSG 1995; Jacobs 1996; PRISMS 1998). The mechanisms of action of IFN‐beta 1b and 1a are not completely understood. Potential mechanisms include regulation of immune cell activation and proliferation, auto‐reactive T‐cell apoptosis, induction of anti‐inflammatory cytokine shifts, inhibition of immune cell trafficking across the blood‐brain barrier and antiviral activity (Dhib‐Jalbut 2010).
GA showed significant efficacy in people with RRMS, reducing the relapse rate and MRI disease activity (Comi 2001; Johnson 1995). The efficacy of GA is considered due to inhibition of the immune response to myelin basic protein and possibly other myelin antigens. Generating suppressor cells, inducing tolerance, expanding regulatory T‐cell populations and altering antigen‐presenting cells seem to be involved in the mechanism of action (Racke 2010).
Why it is important to do this review
The choice of specific DMTs remains a relevant issue, and the final decision should be based on a thorough evaluation of the risk/benefit profile, impact on quality of life and potential neuroprotective and long‐term effects of a given drug (Compston 2008). At present, no clear evidence can be found on the relative efficacy of different DMTs in the treatment of people with RRMS. Comparative trials remain the best tool for acquiring objective information; however, they are difficult to run for several reasons, including the difficulty of achieving proper blinding and the need for large sample sizes (Goodin 2008a).
Although new approved drugs are currently available, IFNs and GA are still used worldwide to treat MS (Jelinek 2015), drug‐persistence is considerable and patients may also resume injectable DMT after suspension for different reasons (Zhornitsky 2015). Specific details on their relative efficacy and tolerability may help physicians make a more precise and unbiased therapeutic choice for their patients.
Dose‐dependent clinical and MRI efficacy of IFNs in RRMS treatment has been suggested, but this study did not analyse the role of GA (Oliver 2010). Differences in the safety profile of immunomodulatory treatments were observed, in particular more favourable skin tolerance with intramuscularly administered IFNs‐beta and a better systemic adverse event profile for GA (Ouallet 2010).
Direct comparative data evaluating IFNs‐beta and GA for RRMS are now available (Goodin 2008b), but no systematic reviews of head‐to‐head trials of IFNs versus GA have been performed.
Objectives
To assess whether IFNs‐beta and GA differ in terms of efficacy and safety in the treatment of people with relapsing‐remitting (RR) MS.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised, double‐blind or single‐blind and open‐label active control trials, as well as cross‐over trials, comparing all types of IFNs versus GA in participants with relapsing‐remitting multiple sclerosis (RRMS). Studies including participants with secondary progressive multiple sclerosis (SPMS) were excluded.
Quasi‐randomised and cross‐over trials were acceptable but were not found. Randomised controlled trials (RCTs) designed with multiple groups were included only with regard to data provided by groups given GA and IFNs. Uncontrolled non‐randomised trials, add‐on trials (i.e. trials with drug associations) and observational studies were excluded. Trials with a follow‐up period shorter than three months were excluded.
Trials comparing head‐to‐head different types and dosages of IFNs without a GA arm were excluded.
Types of participants
People of any age, gender and race affected by RRMS according to Poser 1983's or McDonald's criteria were included (McDonald 2001; Polman 2005; Polman 2011).
A relapsing‐remitting course is characterised by relapses and remissions, with or without complete recovery between relapses.
Study participants had to have an Expanded Disability Status Scale (EDSS) score of 0 to 6.0 (Kurtzke 1983).
Types of interventions
We included trials in which participants received recombinant IFN‐beta 1a (Rebif®, Avonex®) or IFN‐beta 1b (Betaferon®, Betaseron®, Extavia®) at any dose and by any route of administration in any setting.
Comparison: GA at any dose, route of administration and setting.
For trials comparing multiple groups of participants, only the following designs were considered.
-
IFN‐beta 1a versus IFN‐beta 1b versus GA.
-
GA (dose 1) versus GA (dose 2) versus IFNs‐beta.
-
GA versus IFNs‐beta (dose 1) versus IFNs‐beta (dose 2).
Treatment duration had to be at least three months.
Types of outcome measures
Primary outcomes
CLINICAL EFFICACY OUTCOMES
1. Number of participants who experienced at least one relapse at 12 to 24 months and at the end of follow‐up.
2. Number of participants whose condition worsened during the study; we defined worsening as a 0.5‐point increase from starting EDSS score equal to or greater than 5.5; or a 1‐point increase from starting EDSS score equal to or less than 5.0 (Kurtzke 1983). Worsening must have been confirmed during two subsequent neurological examinations separated by at least six months free of relapses (Rudick 2010). We attempted to ascertain confirmed worsening at 12 to 18 months; at 24 months; and at the end of follow‐up. Less stringent criteria (such as an increase in EDSS score sustained for three months) were considered.
CLINICAL SAFETY OUTCOMES
3. Number of participants who withdrew from or dropped out of the study because of adverse events (AEs), defined according to the Cochrane Handbook for Systematic Reviews of Interventions as unfavourable outcomes that occur during or after use of a drug but not necessarily caused by it, and/or as ‘side effects’ (any unintended effects, adverse or beneficial, of a drug that occur at doses normally used for treatment) (Higgins 2011). Serious adverse events (SAEs) were included and were defined according to the US Food and Drug Administration (FDA) as participant outcomes such as death, life‐threatening events, hospitalisation, disability, permanent damage or congenital anomaly/birth defect (FDA 2013).
Secondary outcomes
CLINICAL OUTCOMES
4. Frequency of relapse (number of relapses/patient‐year: annual relapse rate (ARR) was defined as the total number of relapses across all participants divided by total time on the study across all participants). Relapse was defined as newly developed or recently worsened symptoms of neurological dysfunction, with objective confirmation, lasting longer than 24 hours. Less stringent criteria (i.e. without objective confirmation) were considered.
5. Time to first relapse after the start of the study.
6. Percentage of participants free of disease activity: no relapses, no change in EDSS and no MRI changes (T1‐T2).
7. Number of participants treated with steroids for relapse of MS.
8. Mean changes in quality of life (QOL) measured by validated questionnaires such as the Multiple Sclerosis Quality of Life‐54 instrument (MSQOL‐54) (Vickrey 1995).
MRI OUTCOMES
9. Mean number of active (new or enlarged) T2‐hyperintense lesions per participant at 6 ‐12 ‐ 24 months from the start of the study and at the end of the scheduled follow‐up period.
10. Mean number of new contrast‐enhancing T1 lesions per participant at 6 ‐12 ‐ 24 months from the start of the study and at the end of the scheduled follow‐up period.
11. Mean change in total T2‐hyperintense lesion volume at 12 ‐ 24 months from the start of the study and at the end of the scheduled follow‐up period.
12. Mean change in total T1‐hypointense lesion volume at 12 ‐ 24 months from the start of the study and at the end of the scheduled follow‐up period.
13. Mean change in total brain volume (as a measure of atrophy) at 12 ‐ 24 months from the start of the study and at the end of the scheduled follow‐up period.
Search methods for identification of studies
A systematic search with no restrictions was conducted to identify all relevant published and unpublished RCTs.
Electronic searches
The Information Specialist searched the Trials Register of the Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group (last search on 8 August 2016), which, among other sources, contains trials from:
-
The Cochrane Central Register of Controlled Trials (CENTRAL) (8 August 2016);
-
MEDLINE (PubMed) (1966 to 08 August 2016);
-
Embase (Embase.com) (1974 to 8 August 2016);
-
Cumulative Index to Nursing and Allied Health Literature (CINAHL) (EBSCO host) (1981 to 8 August 2016); and
-
Latin American and Caribbean Health Science Information Database (LILACS) (Bireme) (1982 to 8 August 2016).
We searched the following trials registries:
-
Clinical trial registries (clinicaltrials.gov, clinicaltrialsregister.eu); and
-
World Health Organization (WHO) International Clinical Trials Registry Portal (apps.who.int/trialsearch/).
Information on the Trials Register of the Review Group and details of search strategies used to identify trials can be found in the 'Specialised Register' section within the Cochrane Multiple Sclerosis and Rare Diseases of the Central Nervous System Group module.
The keywords used to search for studies for this review are listed in Appendix 1.
Searching other resources
The search was extended to other resources, including:
-
screening of reference lists of review articles and primary studies found;
-
screening of abstract books of the main MS meetings (European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), European Neurological Society (ENS), American Academy of Neurology (AAN) from 2000 to 8 August 2016; and
-
contact with drug manufacturers.
Data collection and analysis
Selection of studies
Three review authors (SF, AG, LLM) independently reviewed titles and abstracts and excluded the study if it did not meet eligibility criteria. They obtained the full paper if the title and the abstract did not provide sufficient information and independently evaluated it. If they could not reach consensus on inclusion/exclusion criteria of an individual study, the final decision was made by all review authors. Study authors were contacted in cases of ambiguity or missing data.
Data extraction and management
For the update of the review, two review authors (AV, AL) independently screened titles and abstracts of the citations retrieved by the literature search to determine their inclusion/exclusion in the review. A third review author (LLM) further revised and confirmed the study selection. The two review authors (AL, LLM) resolved disagreements by discussion.
Assessment of risk of bias in included studies
Two review authors (AV, SF) independently graded the selected trials according to the domain‐based evaluation described in chapter 10 of theCochrane Handbook for Systematic Reviews of Interventions (Sterne 2011). The review authors compared evaluations and discussed and resolved disagreements.
Review authors assessed the following domains as 'yes' (i.e. low risk of bias), 'unclear' (uncertain risk of bias) or 'no' (i.e. high risk of bias).
-
Sequence generation.
-
Allocation concealment.
-
Blinding (of participants, personnel and outcome assessors).
-
Incomplete outcome data (when rate of dropout or loss to follow‐up was greater than 20%, the risk was judged as high).
-
Selective outcome reporting.
The review authors reported on each of these assessments for each individual study in the Risk of bias in included studies table.
We considered overall quality of the studies as good if all domains of selection, attrition and detection were at low risk of bias; moderate if one was at high risk; and poor in the other cases.
Measures of treatment effect
We processed data according to a modified intention‐to‐treat (ITT) principle, using the number of randomly assigned participants who took at least the first dose of the drug. For dichotomous data, we summarised study results as risk ratios (RRs) with 95% confidence intervals (CIs) according to Mantel‐Haenszel methods. When possible, we calculated the number needed to treat for an additional beneficial outcome or the number needed to treat for an additional harmful outcome. For continuous outcomes, weighted mean difference (MD) and standardised mean difference (95% CI) methods were used.
When event rates were reported as the occurrence of events in the overall population over a specific interval in time, we used log‐RR, and when the events were reported as hazard ratio (HR), we used log‐HR.
Unit of analysis issues
Studies with parallel‐group design were included: participants randomly assigned to intervention or control were analysed at the individual allocation level. We decided to include cross‐over studies by considering only data from the first half of the cross‐over trial, but no cross‐over studies were found. We performed a separate analysis at various periods (time frames: short term ≤18 months, medium term 24 and 36 months, and long term > 36 months) of different outcomes based on different periods of follow‐up.
Dealing with missing data
We addressed the effects of withdrawal and loss to follow‐up by performing a sensitivity analysis (see Sensitivity analysis section). When we discovered that some data with focus on predefined outcomes were missing, we contacted the corresponding authors. Missing data are provided in the studies tables. Additional data from the BEYOND trial were provided by Bayer (O'Connor 2009a; Pleimes 2013); but requests for additional data from the other study authors (Lublin 2013a, Mikol 2008 and Calabrese 2012) and from the drug company Merck Serono were not answered.
Assessment of heterogeneity
Clinical diversity and methodological diversity have been considered as heterogeneity. Heterogeneity among trial results has been examined by using the I² test (Higgins 2003). This provides an estimate of the percentage of variability due to heterogeneity rather than to chance alone. We interpreted an I² estimate of 50% or more as indicating the presence of heterogeneity, and random‐effects models were applied (DerSimonian 1986). We investigated diversity in clinical and methodological aspects of included trials.
Assessment of reporting biases
The small number of trials included in this review did not permit an assessment of publication bias. In future updates, we will assess publication bias by following recommendations provided by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Data synthesis
The meta‐analysis was conducted using Review Manager 5 software (Review Manager 2016). If significant heterogeneity was not found among the included trials, we aggregated all included data in the final analyses by using a fixed‐effect model (Yusuf 1985). If substantial clinical diversity was noted between included studies, we used the random‐effects model with studies grouped by intervention. However, we decided to present results using a random‐effects model.
Subgroup analysis and investigation of heterogeneity
Small numbers of trials and small quantities of data did not permit a subgroup analysis according to IFN type and baseline EDSS. In future updates, and if further data become available, we plan to carry out subgroup analyses for primary outcomes according to IFN types beta 1a and 1b and baseline EDSS higher or lower than 3.0 points.
Sensitivity analysis
Sensitivity analyses were performed to explore missing data by likely scenario, attributing the outcome of interest to both treatment groups, as described in the Cochrane Handbook for Systematic Reviews of Interventions, Section 16.2.2 (Higgins 2011).
Summary of findings table
These outcomes are included in the 'Summary of findings' table.
1. Number of participants who experienced at least one relapse.
2. Number of participants whose condition worsened during the study.
3. Number of participants who withdrew from or dropped out of the study because of adverse events.
4. Mean number of active (new or enlarging) T2 lesions.
5. Mean number of new contrast‐enhancing T1 lesions.
6. Mean change in total T2‐hyperintense lesion volume.
7. Mean change in total T1‐hypointense lesion volume.
We assessed the quality of evidence as it relates to the studies that contributed data to the meta‐analyses for prespecified outcomes using GRADEpro software (GRADEpro 2008). We justified all decisions to downgrade or upgrade the quality of studies in footnotes, and we provided comments to aid readers' understanding of the review when necessary, as recommended by Cochrane (Schünemann 2011). The 'Summary of findings' table includes overall grading of the quality of evidence related to each of the outcomes, using the GRADEpro approach (GRADE Working Group 2004). Quality of evidence was graded as high, moderate, low or very low, upon consideration of within‐study risk of bias, directness of evidence, heterogeneity, precision of effect estimates and risk of publication bias. Control event rates used in the calculation of absolute risks were based on the numbers of events reported in the included studies.
Results
Description of studies
Results of the search
We identified overall 636 reports through the search strategy (MEDLINE 356, Embase 225, CINAHL 14, CENTRAL 14, clinical trials registries 11, CDR database 7, other databases 2, additional articles 7). We excluded 592 articles on the basis of abstracts considered not pertinent.
As compared with the first version, we have retrieved 232 new records. Two articles and two new ongoing trials were considered as potentially relevant. One already cited ongoing trial was included in the present updated review (NCT01058005); and one study previously classified as awaiting classification was excluded (Salama 2003). However, the final analysis was unchanged.
Overall, a total of 44 articles were provisionally selected as potentially fulfilling the inclusion criteria (Figure 1).
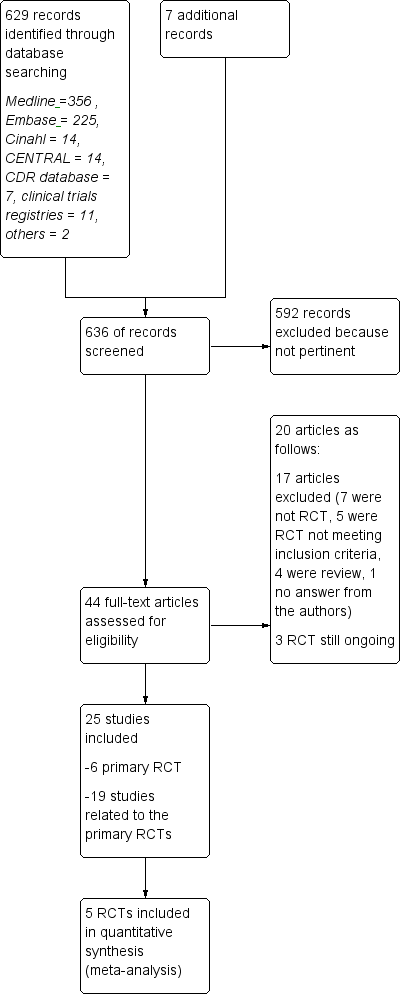
Study flow diagram.
Seventeen studies were excluded (see Characteristics of excluded studies): seven because they were not RCTs (Beer 2011; Carra 2008; Ghezzi 2005; Kalincik 2015; Khan 2001; Khan 2012; Spelman 2015); five RCTs, two on GA because the design included no active comparator treatment with IFNs but placebo in CIS (Comi 2009), or RRMS (Khan 2013), one because it was a comparative trial evaluating two dosages of GA (Comi 2011), two because they evaluated drugs different from those considered in this review (Barbato 2011; Khoury 2010); and four publications because they were review articles (Carter 2010; Del Santo 2011; Ouallet 2010; Qizilbash 2012). Finally one study was excluded because authors never answered our request for additional information (Salama 2003).
Three trials are still ongoing (Characteristics of ongoing studies). One ongoing trial is a phase IV RCT, with head‐to‐head comparisons of IFN‐beta 1b (250 mg of Betaseron) and GA (Copaxone) for the treatment of participants with CIS and RR forms of MS by using acute changes on MRI as the primary outcome (NCT00176592). The second one is a phase IIb/III trial that compares masitinib with interferon‐beta 1a, interferon‐beta 1b, peginterferon‐beta 1a or glatiramer acetate in people with RRMS who do not respond to these first line treatments (EUCTR2012‐003735‐32‐GR). The third is a 12‐month study where participants with RRMS are randomized 1:1 to fingolimod or approved disease modifying therapy (NCT01623596). Participants will be treatment naive or will have only been treated with one class of DMT (Interferon‐beta preparation or glatiramer acetate). The primary objective will be to evaluate efficacy of fingolimod by assessing participants' retention on treatment.
Twenty‐five studies were considered for inclusion: five published RCTs (Cadavid 2009a; Calabrese 2012; Lublin 2013a; Mikol 2008; O'Connor 2009a), and one unpublished study — NCT01058005 — met the selection criteria. Nineteen articles were related to primary studies, excluding the aforementioned ongoing trial (NCT00176592), which was related to the Cadavid 2009a study.
-
Three articles were pertinent to the Cadavid 2009a trial. Investigators analysed the impact of therapies on MRI measures as follows: one was a post hoc analysis of MRI/clinical activity (Cadavid 2011); another was an analysis of the development of focal lesions suggestive of brain injury (Cadavid 2009b); and the last described changes in brain volume (Cheriyan 2012).
-
Six articles were pertinent to the Lublin 2013a trial: one study reported the protocol (Lindsey 2012), and five articles were related to meeting reports of the same trial (Lublin 2012; Lublin 2013b; Lublin 2013c; Wolinsky 2012; Wolinsky 2013).
-
Two articles were pertinent to the Mikol 2008 trial: one was a post hoc analysis of tolerability (Coyle 2010), and the other commented on the same trial (Sørensen 2008).
-
Eight articles were pertinent to the O'Connor 2009a trial: one study was a meeting report of preliminary results (O'Connor 2008); another described the immunological effects of treatments (Goodin 2012); another was a post hoc analysis evaluating MRI measures of degeneration among participants included in the BEYOND trial (Filippi 2011); three were Errata reports (O'Connor 2009b; O'Connor 2011; O'Connor 2012); one was a post hoc analysis (Lampi 2013), and we analysed additional data from the BEYOND trial, as provided by Bayer (Pleimes 2013).
Included studies
Six RCTs met our predefined selection criteria: two studies compared the effects of GA versus IFN‐beta 1b (Cadavid 2009a; O'Connor 2009a), and four compared GA versus IFN‐beta 1a (Calabrese 2012; Lublin 2013a; Mikol 2008; NCT01058005), with two comparing GA versus IFN‐beta 1a 44 mcg SC (Mikol 2008; NCT01058005), one GA versus IFN‐beta 1a 30 mcg IM (Lublin 2013a), one GA versus both IFN‐beta 1a 44 mcg SC and IFN‐beta 1a 30 mcg IM (Calabrese 2012), and one comparing GA versus IFN‐beta 1a 44 mcg SC and natalizumab (NCT01058005)
-
Cadavid 2009a (BECOME) evaluated the efficacy of IFN‐beta 1b and GA in 79 participants with RRMS or CIS (36 treated with IFN and 39 with GA). The primary outcome was MRI measures of activity (combined active lesion counts).
-
Calabrese 2012 evaluated the efficacy of IFN‐beta 1a (44 mcg SC three times weekly), IM IFN‐beta 1a (30 mcg weekly) or GA in 165 participants with RRMS (55 participants in each group). The primary outcome was the development of new cortical lesions and cortical atrophy progression among participants with RRMS.
-
Lublin 2013a (CombiRx) evaluated the efficacy of combined use of interferon‐beta 1a 30 mcg IM weekly and GA 20 mg daily versus each single agent with matching placebo in 1008 participants with RRMS (250 treated with IFN, 259 with GA and 499 with IFN + GA). The primary outcome was annualised relapse rate (ARR).
-
Mikol 2008 (REGARD) evaluated the efficacy of SC IFN‐beta 1a (44 mcg three times weekly) versus GA in 764 participants with RRMS (386 in IFN group and 378 in GA group). The primary outcome was time to first relapse.
-
O'Connor 2009a (BEYOND) evaluated the efficacy of IFN‐beta 1b at two different doses (250 mcg and 500 mcg every other day) and of GA in 2244 participants with RRMS (897 treated with IFN 250 mcg, 899 with IFN 500 mcg and 448 with GA). The primary outcome was risk of relapse. Missing data were provided by Bayer (Pleimes 2013).
-
NCT01058005 (SURPASS) evaluated the safety of natalizumab (300 mg intravenous injection every 4 weeks), IFN‐beta 1a (44 mcg SC injection 3 times weekly), or GA (20 mg SC injection once daily) in 84 participants. The primary outcome was the incidence of treatment‐emergent serious adverse events (SAEs).
The Characteristics of included studies table provides trial design, main characteristics of participants, intervention and treatment groups, selected outcome criteria and the authors' conclusions.
Detailed data related to the definition of relapse are reported in the Characteristics of included studies table. Four of the five trials — Cadavid 2009a; Lindsey 2012; Mikol 2008; O'Connor 2009a — defined relapse as new or recurrent neurological abnormalities associated with an increase in Scripps Neurological Rating Scale (SNRS) score (Cadavid 2009a); or an increase in EDSS score (Cadavid 2009a; Lublin 2013a; Mikol 2008; O'Connor 2009a), lasting at least 24 hours (Cadavid 2009a; Lublin 2013a; O'Connor 2009a), or at least 48 hours (Mikol 2008) and without fever or infection.
NCT01058005 (SURPASS) underwent an early termination due to significantly slower than expected enrolment: all clinical efficacy and magnetic resonance imaging (MRI) procedures were removed from the protocol and safety assessments were to be managed through standard of care activities.The results were reported on clinicaltrials.gov. Based on these data (accessed 19 August 2016) this study was selected because it fitted our predefined inclusion criteria, although it was excluded in the previous version of this review. Actually, no published data have been retrieved and this study did not contribute to any analyses for the lack of available data (Characteristics of included studies).
Cadavid 2009a and Lublin 2013a specified that the increase in EDSS score identified a relapse as follows.
-
Increase in total EDSS of 0.5 point.
-
Increase in Kurtzke Functional System Score (FSS) of 2.0 points.
-
Increase in two or more Kurtzke FSS scores of 1.0 point, or decrease in SNRS score of 7.0 points (Cadavid 2009a).
Disability progression was analysed by four RCTs and was defined as:
-
six months' sustained increase in EDSS ≥ 1.0 point from baseline or at least 0.2 standard deviations (SDs) on baseline Multiple Sclerosis Functional Composite (MSFC) score (post hoc analysis) (Cadavid 2011);
-
six months' sustained increase in EDSS ≥ 1.0 point (0.5 for baseline EDSS ≥ 5.5) (secondary outcome) (Lublin 2013a);
-
six months' sustained increase was confirmed as follows: if the EDSS score at baseline was 0, then a change of 1.5 points or more was required; if the EDSS was 0.5−4.5 at baseline, then a change of 1.0 point or more was required; and if the EDSS at baseline was 5 points or more, then the change required was 0.5 points or more (Mikol 2008);
-
three months' sustained increase in EDSS ≥ 1.0 point (secondary outcome) (O'Connor 2009a).
Baseline characteristics of the included population are reported in Table 1. All RCTs included participants with RRMS with low disability and active disease (frequency of relapse ≥ 1/y). No significant differences were found between studied populations in terms of mean age (range 34.8 to 39.0 years), mean EDSS (1.9 to 2.35 points) and mean relapse frequency (0.97 to 1.9). Mean disease duration was different, ranging from 0.9 (Cadavid 2009a) to 6.55 (Mikol 2008).
| Study name | Drugs | Number of participants | % female | Age, years, mean (SD) | Mean EDSS score (SD) | Disease duration, years,mean (SD) | Previous number of relapses, mean (SD) | Number of participants with MRI Gad+ lesions (%) |
| IFN‐beta 1b | 36 | 75 | 36 (7.75) | 2.0* (0 to 5) | 0.9* (0.1 to 24) | 1.8* (0 to 7.5) | 26 (72) | |
| GA | 39 | 64 | 36 (8.25) | 2.0* (0 to 5.5) | 1.2* (0.2 to 34) | 1,9* (0.13 to 7.0) | 27 (69) | |
| IFN‐beta 1a 44 | 55 | 69.5 | 35.9 (9.1) | 1.9 (1.0) | 5.7 (4.9) | 1,2 (0.6) | ND | |
| IFN‐beta 1a 30 μg | 55 | 68.0 | 34.8 (9.6) | 1.9 (0.8) | 5.3 (5.1) | 1,2 (0.7) | ND | |
| GA | 55 | 72.9 | 38.9 (10.2) | 2.1 (1.1) | 5.5 (6.1) | 1,3 (0.7) | ND | |
| IFN‐beta 1a 30 μg | 250 | 69.2 | 37.6 (10.2) | 2.0 (1.2) | 1.4 (4.0) | 1.7° (0.9) | 187 (74.8) | |
| GA | 259 | 71.4 | 39.0 (9.5) | 1.9 (1.2) | 1.0 (2.9) | 1.6° (0.7) | 215 (83.01) | |
| IFN‐beta 1a 44 μg | 386 | 69 | 36.7 (9.8) | 2.35 (1.28) | 5.93 (6.25) | 0.97** (0.42) | 150 (39) | |
| GA | 378 | 72 | 36.8 (9.5) | 2.33 (1.31) | 6.55 (7.10) | 1.01** (0.35) | 156 (41) | |
| IFN‐beta 1b | 897 | 70 | 35.8** (11.13) | 2.35 (2) | 5.3** (4.45) | 1.6° (0.74) | ND | |
| GA | 448 | 68 | 35.2** (11.87) | 2.28 (2) | 5.1** (4.45) | 1.6° (0.74) | ND |
ND (no data available).
*Median (range); mean not reported.
**SD or weighted mean was calculated.
°Pre‐1‐year.
Only three studies reported the numbers of participants with enhancing baseline lesions at MRI (Cadavid 2009a; Lublin 2013a; Mikol 2008).
The overall number of participants included in the five RCTs was 4256. However, we decided to exclude from the analysis high dosages of IFN‐beta 1b (500 mcg to 899 participants), as used in the O'Connor 2009a study, and combined IFN + GA (509 participants), as used in Lublin 2013a study arms, because these schedules are not used in clinical practice. The overall population considered in our analysis was 2858 (1679 participants treated with IFN and 1179 with GA). The drugs analysed in comparison with GA were IFN‐beta 1b (two trials, 933 participants), SC IFN‐beta 1a 44 mcg (two trials, 441 participants) and IM IFN‐beta 1a 30 mcg (two trials, 305 participants).
Duration of treatment and follow‐up was three years for the Lublin 2013a study and two years for the other four RCTs.
Excluded studies
See 'Results of the search' and the Characteristics of excluded studies table.
Risk of bias in included studies
The risk of bias was variable across studies: incomplete outcome data was the main biased dimension (high risk of bias in all studies) because of high levels of dropout and missing data, followed by lack of blinding of participants and investigators and by selective outcome reporting (high risk of bias in three and two studies, respectively).
Further details of this assessment are provided in relevant sections of the Characteristics of included studies table and are presented as the 'Risk of bias' graph (Figure 2) and the 'Risk of bias' summary (Figure 3).
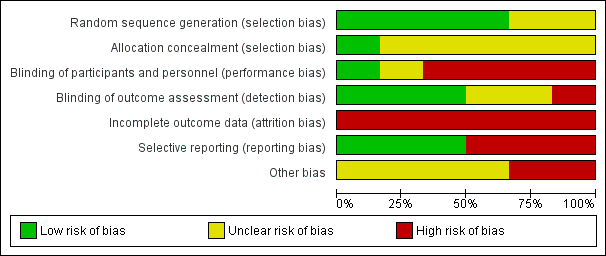
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies, with the exception of Cadavid 2009a (in which the item was not mentioned) used computer systems to generate the allocation sequence, but none clearly explained how the sequence was concealed (with the exception of Lublin 2013a, in which the sequence was masked by the computer system).
Blinding
Participants treated in Mikol 2008, O'Connor 2009a, Cadavid 2009a and NCT01058005 were not blinded. In Calabrese 2012, this item is not mentioned and in Lublin 2013a the computer system ensured blindness. Treating physicians were aware of the treatments in Mikol 2008 and O'Connor 2009a and were unaware in Lublin 2013a. The other studies did not mention this item. In all studies, outcome assessors were blinded. Two studies were at low risk for detection bias for MRI measures (Cadavid 2009a; Calabrese 2012), which were the primary outcomes for these studies and were unclear for clinical outcomes.
Incomplete outcome data
Incomplete outcome data was the main biased dimension because of the high level of loss to follow‐up. Participants who dropped out accounted for 13% to 30%, with higher values for IFN groups in four RCTs (Table 2). An ITT analysis was performed in Cadavid 2009a, Mikol 2008 and Lublin 2013a, but in the other two studies it was not performed. Reasons for loss to follow‐up were clearly reported only by Cadavid 2009a and Mikol 2008.
| Study | Arm | N° participants | N° dropout | Reasons for missing | |||||||||||
| At randomisation | 24 months | 36 months | n° | % | Lost to follow‐up | Did not receive drug | Switched treatment | Withdrew | Deviated from protocol | Had adverse events | Died | Became pregnant | Other reasons | ||
| IFN‐beta 1a 44 μg | 36 | 25 | ND | 116 | 30.6 | 7 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 38 | |
| GA | 39 | 31 | ND | 87 | 20.5 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 48 | |
| IFN‐beta 1a 44 μg | 55 | 46 | ND | 9 | 16.4 | 9 | ND | ||||||||
| IFN‐beta 1a 30 μg | 55 | 47 | ND | 8 | 14.5 | 8 | |||||||||
| GA | 55 | 48 | ND | 7 | 12.7 | 7 | |||||||||
| IFN‐beta 1a 30 μg | 250 | ND | 194 | 56 | 22.4 | 13 | 0 | 0 | 0 | 0 | 1711 | 1 | 0 | 2513 | |
| GA | 259 | ND | 223 | 36 | 13.9 | 9 | 0 | 0 | 0 | 0 | 1112 | 1 | 0 | 1514 | |
| IFN‐beta 1a 44 μg | 386 | 301 | ND | 85 | 22.0 | 17 | 3 | 0 | 0 | 2 | 23 | 0 | 8 | 329 | |
| GA | 378 | 324 | ND | 54 | 14.3 | 2 | 3 | 0 | 0 | 2 | 19 | 0 | 5 | 2310 | |
| IFN‐beta 1b 250 | 897 | 784 | ND | 113 | 12.6 | 12 | 9 | 10 | 38 | 3 | 13 | 1 | 27 | ||
| GA | 448 | 374 | ND | 74 | 16.5 | 12 | 3 | 5 | 18 | 2 | 8 | 1 | 1 | 24 | |
ND (no data available)
Source of data are described as follow.
1] pg 1977; 2] pg 3; 3] pg 33; 4] pg 904; 5] pg 890; 6] 7 lost to follow‐up + 4 discontinued intervention (see pg 1977); 7] 4 lost to follow‐up + 4 discontinued intervention (see pg 1977); 8] treatment failure; 9] 4 had disease progression + 28 for other reasons; 10] 7 had disease progression + 16 for other reasons; 11] 4 AE/SAE + 13 side effects; 12] 6 AE/SAE + 5 side effects; 13] 14 non‐medical reason + 11 other medical reason; 14] 8 non‐medical reason + 7 other medical reason
The online publication was corrected after the first publication of the study became available (O'Connor 2009b; O'Connor 2011; O'Connor 2012).
Selective reporting
Cadavid 2009a and O'Connor 2009a failed in matching outcomes planned with outcomes reported: O'Connor 2009a reported five tertiary outcomes not planned, and Cadavid 2009a reported one outcome more than those planned.
Other potential sources of bias
All studies were sponsored by the drug industry (Bayer sponsored Cadavid 2009a and O'Connor 2009a; Merck Serono sponsored Mikol 2008 and Calabrese 2012), with the exception of Lublin 2013a, which was funded by the National Institutes of Health and the National Institute of Neurological Disorders and Stroke.
Effects of interventions
Main findings are described in summary of findings Table for the main comparison.
Clinical efficacy outcomes
1. Number of participants who experienced at least one relapse at 12 months or at 24 months or at the end of follow‐up
This outcome was assessed by three trials at 24 months (Cadavid 2009a; Mikol 2008; O'Connor 2009a) (2184 participants; 76%) and by one trial at 36 months (Lublin 2013a) (509 participants; 18%) and was not available from the Calabrese 2012 trial. From these data, we found no significant differences in effect at 24 months (RR 1.04, 95% CI 0.87 to 1.24) and at 36 months (RR 1.27, 95% CI 0.92 to 1.75). No significant heterogeneity was found among studies (Analysis 1.1). The results were unchanged when missing data were explored at sensitivity analysis by a likely scenario (Analysis 1.2) (Table 3).
| Study | Arm | N° participants | N° dropout | Lost to follow‐up | |||
| Randomised available for the outcome | 24 months | 36 months | n° | % | |||
| IFN‐beta 1a 44 | 36 | 25 | 11 | 30.6 | 7 | ||
| GA | 39 | 31 | 8 | 20.5 | 4 | ||
| IFN‐beta 1a 44 | 55 | 46 | 9 | 16.4 | 9 | ||
| IFN‐beta 1a 30 | 55 | 47 | 8 | 14.5 | 8 | ||
| GA | 55 | 48 | 7 | 12.7 | 7 | ||
| IFN‐beta 1a 30 | 250 | 194 | 56 | 22.4 | 13 | ||
| GA | 259 | 223 | 36 | 13.9 | 9 | ||
| IFN‐beta 1a 44 | 386 | 301 | 85 | 22.0 | 17 | ||
| GA | 378 | 324 | 54 | 14.3 | 2 | ||
| IFN‐beta 1b | 897 | 784 | 113 | 12.6 | 57* | ||
| GA | 448 | 374 | 74 | 16.5 | 31* | ||
*Data provided by Bayer (Pleimes 2013).
2. Number of participants with confirmed worsening at 12 months or at 24 months or at the end of follow‐up
This outcome was assessed by three trials at 24 months (2169 participants; 76%) and by one trial at 36 months (487 participants; 17%). Three trials adopted the predefined criteria of worsening (confirmed during two subsequent neurological examinations separated by at least six months) (Cadavid 2009a; Mikol 2008,Lublin 2013a). One study used less stringent criteria (i.e. an increase in EDSS sustained for three months) (O'Connor 2009a).
No differences were found when confirmed progression was analysed at 24 months (RR 1.11, 95% CI 0.91 to 1.35) or at 36 months (RR 0.87, 95% CI 0.63 to 1.20) (Analysis 2.1).
Results were unchanged when missing data were explored by a likely scenario (Analysis 2.2) (Table 4).
| Study | Arm | N° participants at | N° dropout | Lost to follow‐up | |||
| Randomised available for the outcome | 24 months | 36 months | n° | % | |||
| IFN‐beta 1a 44 | 36 | 25 | 11 | 30.6 | 7 | ||
| GA | 39 | 31 | 8 | 20.5 | 4 | ||
| IFN‐beta 1a 44 | 55 | 46 | 9 | 16.4 | 9 | ||
| IFN‐beta 1a 30 | 55 | 47 | 8 | 14.5 | 8 | ||
| GA | 55 | 48 | 7 | 12.7 | 7 | ||
| IFN‐beta 1a 30 | 241 | 194 | 47 | 19.5 | 13 | ||
| GA | 246 | 223 | 23 | 9.3 | 9 | ||
| IFN‐beta 1a 44 | 386 | 301 | 85 | 22.0 | 17 | ||
| GA | 378 | 324 | 54 | 14.3 | 2 | ||
| IFN‐beta 1b | 886 | 784 | 102 | 11.5 | 80* | ||
| GA | 444 | 374 | 70 | 15.8 | 58* | ||
*Data provided by Bayer (Pleimes 2013).
Clinical safety outcomes
3. Number of participants who withdrew from or dropped out of the study because of adverse events
The number of participants who withdrew from or dropped out of the study because of adverse events was available for four studies (2685 participants; 93%) and was not reported in the Calabrese 2012 study. No differences were found between the two treatment groups (RR 0.95, 95% CI 0.64 to 1.40). No heterogeneity was found (Analysis 3.1).
Similar results were found when SAEs were considered (RR 0.99, 95% CI 0.63 to 1.56) (Lublin 2013a; Mikol 2008; O'Connor 2009a). Seven deaths were reported: five in the IFN group (three in the 500 mcg arm) and two in the GA group: among participants treated with IFN, one died (suicide) about three months after taking the last dose of study drug (IFN‐beta 1a) (Mikol 2008), one died as the result of pulmonary embolism (Lublin 2013a), and three died for unexplained reasons (O'Connor 2009a). No reason was specified for one death reported in the GA group (O'Connor 2009a), and one participant died of a large cell lymphoma of the CNS (Lublin 2013a) (Analysis 3.2).
Lublin 2013a is the only trial that provided details on numbers of SAEs by organ system as experienced by participants. In this study, SAEs occurred more often in the IFN group than in the GA group: nervous system disorders, including relapses: 4.4% versus 1.9%; neoplasms benign and malignant: 2% versus 0.4%; surgical and medical procedures: 2% versus 1.9%; infections and infestations: 1.6% versus 0.4%; gastrointestinal disorders: 1.2% versus 0.8%; and cardiac disorders: 1.2% versus 0.8%.
By contrast, the proportions of participants treated with GA were higher for experiencing hepatobiliary disorders (0.8% vs 0.4%) and musculoskeletal, connective tissue and bone disorders (0.8% vs 0.4%).
NCT01058005 reported two SAEs: one meningitis herpes and one cerebral venous thrombosis in 2/22 participants treated with IFN1a.
Analysis of the number needed to treat for an additional beneficial outcome or the number needed to treat for an additional harmful outcome was not provided because reliable data were insufficient.
Clinical outcomes
4. Frequency of relapse
The frequency of relapse was analysed in terms of log rate ratio at 24 months for four studies (Cadavid 2009a; Calabrese 2012; Mikol 2008; O'Connor 2009a). The rate ratio (1.06, 95% CI 0.95 to 1.18) showed no difference between the two groups. At 36 months, data were provided by one study (Lublin 2013a); the rate ratio (1.40, 95% CI 1.13 to 1.74) was significantly higher in the IFN group (P value 0.002), favouring GA (Analysis 4.1).
5. Time to first relapse
No differences were found (HR 1.01, 95% CI 0.87 to 1.16) without heterogeneity among studies (Analysis 5.1).
6. Percentage of participants free of disease activity: no relapses, no changes in EDSS and no MRI changes
No analysis was possible because data were insufficient. Data were reported by only one study (Lublin 2013a): a preplanned assessment of percentages of participants with no clinical or MRI activity was performed and no significant differences were found between IFNs and GA (21.2% and 19.4%).
7. Participants treated with steroids for relapse of MS
This outcome was available for two studies (1420 participants; 50%) (Cadavid 2009a; O'Connor 2009a). Results did not show a statistically significant difference between the two therapies (RR 1.30, 95% CI 0.76 to 2.24) (Analysis 7.1). Significant heterogeneity was found between studies (I² = 63%).
8. Mean changes in quality of life
No data were available.
MRI outcomes
9. Mean number of active (new or enlarged) T2‐hyperintense lesions per participant at 6 or 12 or 24 months
This outcome was available for one study at six months (396 participants; 14%), for two studies at 12 months (1722 participants; 60%) and for three studies at 24 months (1790 participants; 62%). For the latter time point, data from Calabrese 2012 referred to the IFN 44 mcg group. At six months, the number of active (new or enlarging) T2 lesions was significantly lower in IFN‐treated than in GA‐treated participants (MD −0.86, 95% CI −1.32 to −0.40; P value 0.0003). No significant differences were found between IFN and GA for this outcome at 12 months or at 24 months (MD −0.52, 95% CI −1.12 to 0.09, and MD −0.15, 95% CI −0.68 to 0.39, respectively). At 24 months, heterogeneity was significant (I² = 68%) (Analysis 9.1). At 24 months, the results did not change when data from Calabrese 2012 evaluating the effects of IM IFN‐beta 1a 30 mcg versus GA were analysed (MD 0.11, 95% CI −0.67 to 0.44).
10. Mean number of new contrast‐enhancing T1 lesions per participant at 6 or 12 or 24 months
This outcome was available at 12 months for one study (1233 participants; 43%) and at 24 months for three studies (1734 participants; 61%). For the latter time point, data from Calabrese 2012 referred to the IFN 44 mcg group. No significant differences between SC IFN 44 mcg and GA were found for this outcome at 12 months (MD −0.10, 95% CI −0.26 to 0.06) and at 24 months (MD −0.14, 95% CI −0.30 to 0.02) (Analysis 10.1). At 24 months, the results did not change when data from Calabrese 2012 evaluating the effects of IM IFN‐beta 1a 30 mcg versus GA were analysed (MD −0.16, 95% CI −0.31 to 0.00).
No data at six months were available.
11. Mean change in total T2‐hyperintense lesion volume at 12 or 24 months
This outcome was available for one study at 12 months (1221 participants; 43%), for two studies at 24 months (1608 participants; 56%) and for another study at 36 months (509 participants; 19%). The mean increase in T2 lesion volume was significantly less in IFN‐treated than in GA‐treated participants at month 12 and at month 24 (MD −0.40, 95% CI −0.59 to −0.21, and MD −0.58, 95% CI −0.99 to −0.18; P values < 0.0001 and 0.004, respectively). No heterogeneity was found. The difference favouring IFN (MD −0.26, 95% CI −1.04 to 0.52) was also present in 36‐month data from a single study (Lublin 2013a), but it did not reach statistical significance (Analysis 11.1).
12. Mean change in total T1‐hypointense lesion volume at 12 or 24 months
This outcome was available at 12 months for one study (1207 participants; 42%) and at 24 months for two studies (1602 participants; 56%). The mean increase in T1 lesion volume was significantly less in IFN‐treated than in GA‐treated participants at month 24 (MD −0.20, 95% CI −0.33 to −0.07; P value 0.003). The difference favouring IFN (MD −0.06, 95% CI −0.18 to 0.07) was also seen in 12‐month data from a single study (O'Connor 2009a), but did not reach statistical significance (Analysis 12.1).
13. Mean change in total brain volume (as a measure of atrophy) at 12 or 24 months
This outcome was available at 12 months for one study (1137 participants; 40%) and at 24 months for two studies (1552 participants; 54%). Data at 12 months did not show a significant difference between IFN and GA with regard to brain volume changes (MD −0.10, 95% CI −0.22 to 0.02). At 24 months, mean brain volume reduction was significantly greater in IFN‐treated than in GA‐treated participants (MD −0.12, 95% CI −0.23 to −0.01; P value 0.04). At 24 months, the heterogeneity was significant (I² = 83%) (Analysis 13.1).
Discussion
Summary of main results
The purpose of this study was to evaluate whether treatment with IFN‐beta and GA in participants with RRMS was different in terms of efficacy and safety, by performing a systematic review of head‐to‐head RCTs.
Six trials were included and five trials contributed to this review with data. The overall population included 2904 participants (1704 participants treated with IFN and 1200 with GA). The drugs analysed in comparison with GA were SC IFN‐beta 1b (two trials, 933 participants), SC IFN‐beta 1a 44 mcg (three trials, 466 participants) and IM IFN‐beta 1a 30 mcg (two trials, 305 participants). Duration of treatment and follow‐up was three years for Lublin 2013a and two years for the other four RCTs. All studies were at high risk for attrition bias.
The main conclusion was that the two therapies seemed to have similar effects in terms of clinical efficacy, when primary outcomes (number of participants with relapse or risk of confirmed progression) and other supportive outcomes (time to first relapse, number of participants treated with steroids) were considered. However, we found some evidence, limited by the small number of participants derived from only one study, that the relapse ratio was significantly lower at 36 months in GA as compared with IFN.
The number of participants who withdrew from or dropped out of the studies because of adverse events (safety end point) was similar in the two groups. The percentage of discontinuation due to adverse effects was similar (3% for IFNs and 4% for GA). The percentage of discontinuation due to serious adverse events was also similar between groups.
In light of MRI outcomes, results showed that the effects on new/enlarging T2 or gadolinium‐enhancing lesions at 24 months were similar. However, IFNs have a more pronounced effect on T2 and T1 lesion volume accrual at 12 (T2) and 24 months. Conversely, a lower rate of brain volume (BV) loss at 24 months was observed in GA‐treated participants. The values for new T2 lesions and for brain atrophy data were limited by the heterogeneity of the results. Methodological issues have been highlighted (Rudick 2009).
The quality of evidence for primary outcomes was judged as moderate for clinical end points, but for safety and some MRI outcomes (number of active T2 lesions) as low (summary of findings Table for the main comparison).
Overall completeness and applicability of evidence
The RCTs included in the review were homogeneous in terms of included populations, treatment schedules and outcome measures.
All studies included only participants with active RRMS (pre‐study relapse frequency ranging from 0.97 to 1.9) and low disability (EDSS 1.9 to 2.35). Participants had similar baseline characteristics in terms of gender and mean age (34.8 to 39 years), although mean disease duration was different, ranging from 0.9 years to 6.55 years (Cadavid 2009a and Mikol 2008, respectively).
Studies compared the effects of different IFN products used in clinical practice versus GA. However, evaluation of the comparative effects of IFN‐beta 1a IM (Avonex) and GA was limited because of the different time points provided (24 months and 36 months, respectively). For this reason, the main comparison in this review was that between GA and IFNs at high frequencies and dosages.
The objectives of this review were reasonably well achieved, allowing comparison of the two treatments in terms of predefined clinical and MRI measures of efficacy. The main outcome measure not available for analysis was quality of life, which impaired the comparison of the relative tolerability of different DMTs in terms of patient‐reported outcomes.
We selected outcome measures that evaluated changes in clinical activity (relapse rate, attack‐free status or time to first relapse), MRI activity (new or enlarging T2 lesions or Gd‐enhancing lesions), clinical severity (confirmed disability progression) or MRI severity (T1 lesion load accrual or BV decrease).
Overall, results showed no differences in clinical effect between the two treatments. Different effects on some measures of MRI activity suggest that IFN might have a stronger and faster anti‐inflammatory action than GA. Such an anti‐inflammatory effect might also account for observed differences between the two drugs in terms of brain volume reduction at 24 months, because they could be related to non–tissue‐related BV loss ('pseudoatrophy' effect), although pseudoatrophy is seen primarily only during the first year (Zivadinov 2008). Furthermore, the clinical impact of these data is currently being studied (Fisniku 2008: Popescu 2013).
The results of this systematic review have several limitations.
-
The main limitation was related to high risk of attrition bias. The percentage of dropouts was higher in the IFN groups than in the GA group for different reasons (adverse effects, lack of consent, tolerability). This aspect might have a role in the final assessment of effectiveness of these drugs, although sensitivity analysis using a likely scenario did not capture different results.
-
The definition of relapse was similar, with objective confirmation required, in all studies. However, Calabrese 2012 did not define the outcome, and Mikol 2008 counted as relapses both qualifying and non‐qualifying attacks. We could not exclude the possibility that an inhomogeneous relapse definition might constitute a bias.
-
Pivotal trials comparing DMTs versus placebo have suggested different efficacy for disease activity (Hillert 2012), although the results of this review show similar effects for IFN and GA. The reasons for this are uncertain, but the primary source of discrepancy is probably related to differences in the enrolled populations of studies, especially in the behaviour of placebo groups (Goodin 2008a).
-
Data did not allow verification of the effects of IFNs and GA as change therapy in participants who were not responders to previous therapies. A recent comparative study evaluating DMTs in real world clinical practice using international MSBase registry data has been conducted using propensity score matching. Slightly lower relapse incidence was found among participants treated with glatiramer acetate or subcutaneous interferon‐beta 1a relative to intramuscular interferon‐beta 1a and interferon‐beta 1b. No differences in 12‐month confirmed progression of disability were observed (Kalincik 2015). The ongoing RCTs might possibly contribute to this issue comparing the new approved therapies.
Analysis of the safety profile was restricted to the number of participants who withdrew from, or dropped out of, the study; drug‐related adverse effects (tolerability) were not considered. Data derived from one study (reported but not planned) suggest a greater incidence of SAEs such as tumour or infection in IFN groups than in GA groups, although hepatobiliary and musculoskeletal disorders were reported more often among GA‐treated participants. Other studies showed that tolerability was generally good. It is well known that up to 75% of IFN‐treated patients experience flu‐like illness, headache and myalgia. Increased alanine aminotransferase and leukopenia occur significantly more often in the IFN group, whereas pruritus, swelling, induration at the injection site (56% to 71%), dyspnoea and post‐injection systemic reactions (15% to 38%) occur significantly more often in the GA group (Carter 2010; Parkenov 2013).
Significant heterogeneity of results on MRI assessment of new or enlarging T2 and brain volume changes was probably related to differences in the MRI protocol and measures of image acquisition protocol (Cadavid 2009a; O'Connor 2009a; Filippi 2011).
Quality of the evidence
Six RCTs were included, but only five contribute to the analyses involving large numbers of participants with similar disease characteristics. No heterogeneity of results was found when clinical end points were considered, supporting no difference in the protective effects of IFNs and GA on risk of relapse and progression.
The body of evidence was judged to be of moderate quality for most clinical and radiological outcomes, although the quality of evidence for the safety profile was found to be low (summary of findings Table for the main comparison).
Potential biases in the review process
The trials search strategy and contacts initiated with the main investigators suggest the likelihood that all relevant studies were identified and all relevant data were obtained.
Agreements and disagreements with other studies or reviews
This is the first systematic review comparing IFNs and GA by using head‐to‐head RCTs in participants with RRMS.
Many reviews have been published evaluating the efficacy and safety of DMTs and other approved therapies through direct or indirect comparison in participants with RRMS, but some have considered different types of trials, outcome measures and designs.
A Cochrane systematic review reported a meta‐analysis of all available therapies in MS, including comparisons between active and placebo groups and head‐to‐head studies (Filippini 2013). Head‐to‐head trials on IFN and GA were also considered. However, in terms of risk of relapse and worsening at 24 months, the quality of the evidence was judged too low to allow meaningful comparisons, given the small sample size and the wide 95% confidence intervals. In this review, only two studies were considered (Mikol 2008; O'Connor 2009a).
Qizilbash 2012 considered both RCTs and comparative observational cohort studies and focused on participants treated with GA for RRMS or CIS versus placebo; the comparators in a subanalysis were also IFNs and standard treatment. Eleven studies were included. As compared to the present study results, an 18% reduction in clinical progression (RR 0.82, 95% CI 0.68 to 0.98) was seen in GA‐treated participants compared with those given IFNs, but only two studies were included (Mikol 2008; O'Connor 2009a). Similarly to our findings, study discontinuations due to adverse events did not differ among participants treated with GA and those given IFNs (RR 0.89, 95% CI 0.57 to 1.41). Hadjigeorgiou 2013 evaluated through direct and indirect comparisons the approved existing treatment for RRMS, but no clear conclusions were provided. Zagmutt 2013 conducted a network meta‐analysis to assess the rates of adverse events and dropout from treatment for RRMS. Placebo‐controlled RCTs evaluating immunosuppressive drugs and DMTs were included. The GA group had a significantly lower incidence rate of adverse events when compared with groups given other products. Dropouts were not significantly different across treatments, except for a higher incidence in the SC IFN‐beta 1a 22 mcg and 44 mcg groups compared with the fingolimod group.
A recent comparative study evaluating DMTs in real world clinical practice using international MSBase registry data has been conducted using propensity score matching: a slightly lower relapse incidence was found among participants treated with glatiramer acetate or subcutaneous interferon‐beta 1a relative to intramuscular interferon‐beta 1a and interferon‐beta 1b. No differences in 12‐month confirmed progression of disability were observed (Kalincik 2015).
A systematic non‐Cohrane review of observational studies and extension of randomized controlled trials (not included in our review) has been conducted to evaluate the long‐term impact of IFNs or GA on disability progression in MS. The quantitative estimate of the treatment effect in reducing progression to EDSS 6 was HR pooled = 0.49 (95% CI 0.34 to 0.69), P < 0.001, showing that treatment with immunomodulators seems to reduce long‐term probability of disability progression (Signori 2016).
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
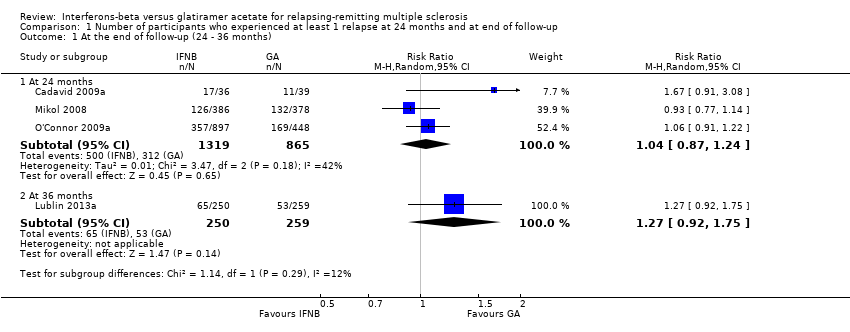
Comparison 1 Number of participants who experienced at least 1 relapse at 24 months and at end of follow‐up, Outcome 1 At the end of follow‐up (24 ‐ 36 months).
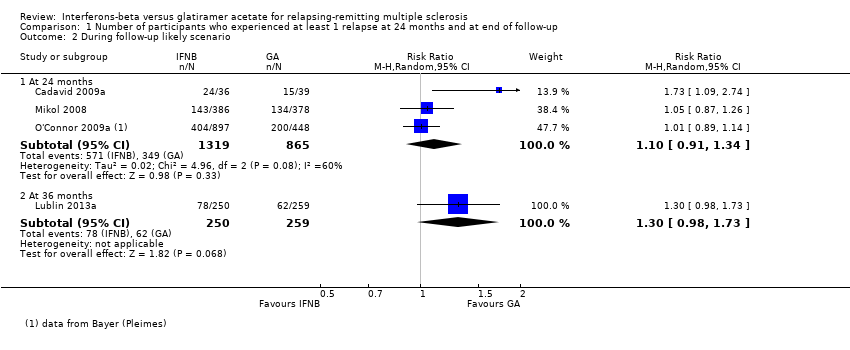
Comparison 1 Number of participants who experienced at least 1 relapse at 24 months and at end of follow‐up, Outcome 2 During follow‐up likely scenario.

Comparison 2 Number of participants who worsened during the study (EDSS progression) at 24 months and at the end of follow‐up, Outcome 1 At the end of follow‐up (24‐36 months).

Comparison 2 Number of participants who worsened during the study (EDSS progression) at 24 months and at the end of follow‐up, Outcome 2 During follow‐up likely scenario.

Comparison 3 Number of participants who withdrew or dropped out of the study because of adverse events, Outcome 1 Number of participants who dropped out for AE.

Comparison 3 Number of participants who withdrew or dropped out of the study because of adverse events, Outcome 2 Number of participants who dropped out for SAE.
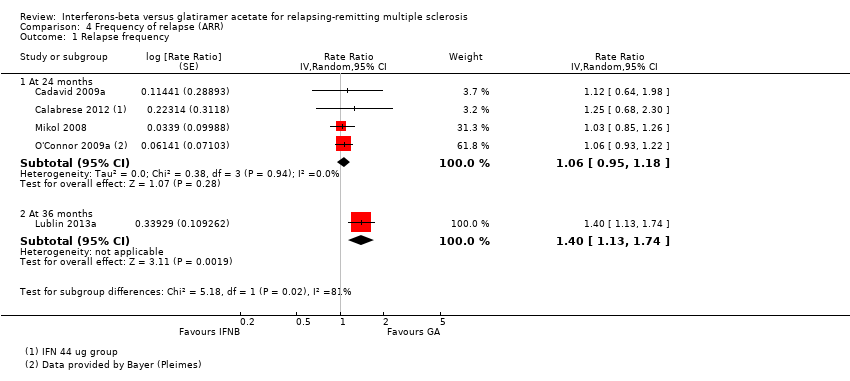
Comparison 4 Frequency of relapse (ARR), Outcome 1 Relapse frequency.
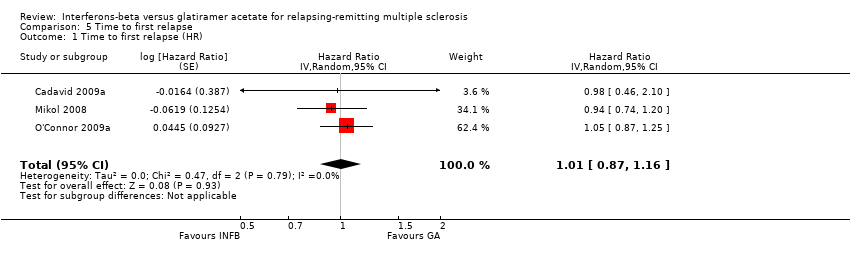
Comparison 5 Time to first relapse, Outcome 1 Time to first relapse (HR).

Comparison 7 Number of participants treated with steroids for MS relapse, Outcome 1 Patients Treated with Steroids.
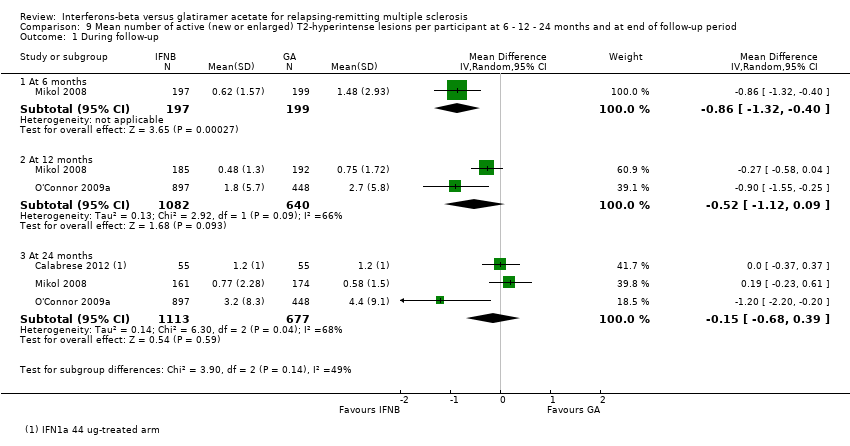
Comparison 9 Mean number of active (new or enlarged) T2‐hyperintense lesions per participant at 6 ‐ 12 ‐ 24 months and at end of follow‐up period, Outcome 1 During follow‐up.
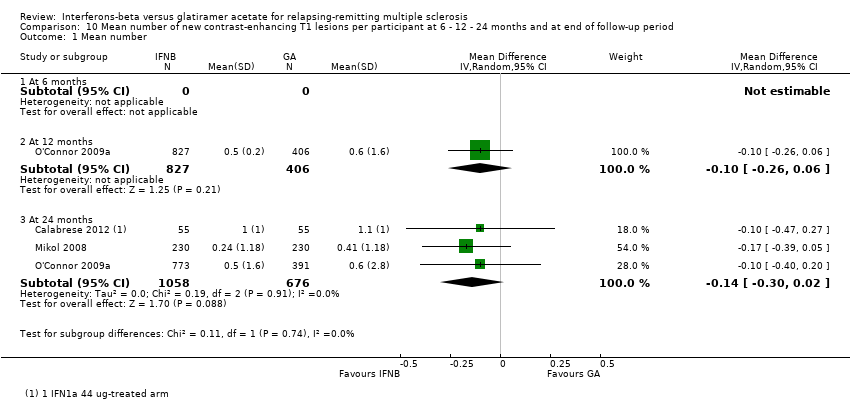
Comparison 10 Mean number of new contrast‐enhancing T1 lesions per participant at 6 ‐ 12 ‐ 24 months and at end of follow‐up period, Outcome 1 Mean number.

Comparison 11 Mean change in total T2‐hyperintense lesion load at 12‐24 months and at end of follow‐up period, Outcome 1 Mean absolute change.
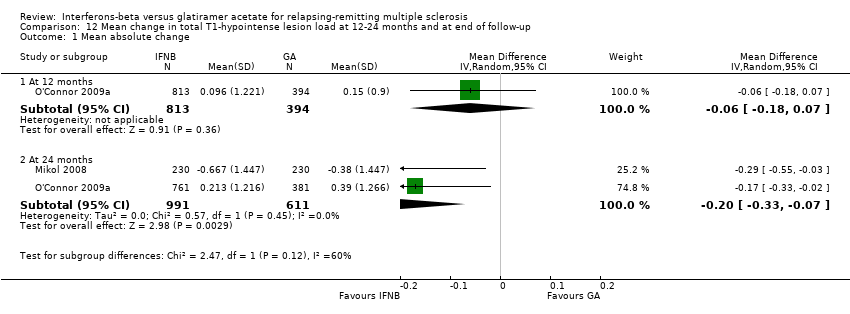
Comparison 12 Mean change in total T1‐hypointense lesion load at 12‐24 months and at end of follow‐up, Outcome 1 Mean absolute change.
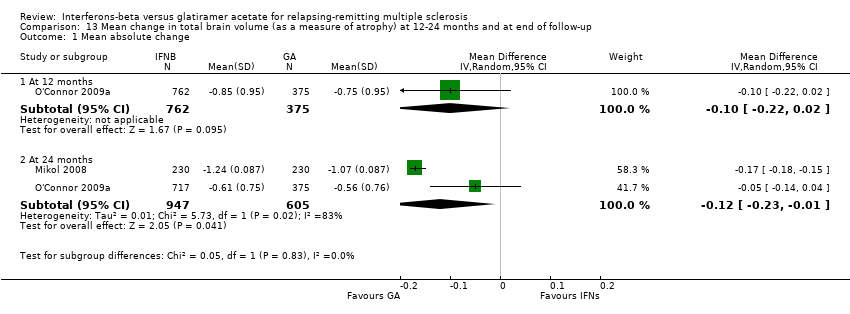
Comparison 13 Mean change in total brain volume (as a measure of atrophy) at 12‐24 months and at end of follow‐up, Outcome 1 Mean absolute change.
| interferons compared with glatiramer acetate for participants with relapsing‐remitting multiple sclerosis | ||||||
| Patient or population: people with relapsing‐remitting multiple sclerosis | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk (control) | Corresponding risk (intervention) | |||||
| Glatiramer acetate | Interferons | |||||
| Number of participants with relapse (M‐H, random, 95% CI) | Study population | RR 1.04 | 2184 | ⊕⊕⊕⊝ | Detection bias risk for clinical outcomes was judged as high for 1 study and low for the other 2 RCTs | |
| 36 per 100 | 38 per 100 | |||||
| Moderate | ||||||
| 35 per 100 | 36 per 100 | |||||
| Number of participants with confirmed progression (M‐H, random, 95% CI) | Study population | RR 1.11 | 2169 | ⊕⊕⊕⊝ | Detection bias risk for clinical outcomes was judged as high for 1 study and low for the other 2 RCTs | |
| 15 per 100 | 16 per 100 | |||||
| Moderate | ||||||
| 15 per 100 | 17 per 100 | |||||
| Number of participants who dropped out for AEs (M‐H, random, 95% CI) | Study population | RR 0.95 | 2685 | ⊕⊕⊝⊝ | ||
| 4 per 100 | 4 per 100 | |||||
| Moderate | ||||||
| 5 per 100 | 5 per 100 | |||||
| Mean number of active T2 lesions | 0.15 lower in IFN versus GA groups | 1790 | ⊕⊕⊝⊝ | Detection bias risk for MRI outcomes was judged as low for all studies | ||
| Mean number of new enhancing lesions | 0.14 lower in IFN versus GA groups | 1734 | ⊕⊕⊕⊝ | Detection bias risk for MRI outcomes was judged as low for all studies | ||
| Mean change in total T2‐hyperintense lesion load | 0.58 lower in IFN versus GA groups | 1608 | ⊕⊕⊕⊝ | Detection bias risk for MRI outcomes was judged as low for both studies | ||
| Mean change in total T1‐hypointense lesion load | −0.20 lower in IFN versus GA groups (−0.33 to −0.07) | 1602 | ⊕⊕⊕⊝ | Detection bias risk for MRI outcomes was judged as low for both studies | ||
| *The basis for the assumed risk (e.g. median control group risk (GA) across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group (IFNs) and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aHigh risk of attrition bias. | ||||||
| Study name | Drugs | Number of participants | % female | Age, years, mean (SD) | Mean EDSS score (SD) | Disease duration, years,mean (SD) | Previous number of relapses, mean (SD) | Number of participants with MRI Gad+ lesions (%) |
| IFN‐beta 1b | 36 | 75 | 36 (7.75) | 2.0* (0 to 5) | 0.9* (0.1 to 24) | 1.8* (0 to 7.5) | 26 (72) | |
| GA | 39 | 64 | 36 (8.25) | 2.0* (0 to 5.5) | 1.2* (0.2 to 34) | 1,9* (0.13 to 7.0) | 27 (69) | |
| IFN‐beta 1a 44 | 55 | 69.5 | 35.9 (9.1) | 1.9 (1.0) | 5.7 (4.9) | 1,2 (0.6) | ND | |
| IFN‐beta 1a 30 μg | 55 | 68.0 | 34.8 (9.6) | 1.9 (0.8) | 5.3 (5.1) | 1,2 (0.7) | ND | |
| GA | 55 | 72.9 | 38.9 (10.2) | 2.1 (1.1) | 5.5 (6.1) | 1,3 (0.7) | ND | |
| IFN‐beta 1a 30 μg | 250 | 69.2 | 37.6 (10.2) | 2.0 (1.2) | 1.4 (4.0) | 1.7° (0.9) | 187 (74.8) | |
| GA | 259 | 71.4 | 39.0 (9.5) | 1.9 (1.2) | 1.0 (2.9) | 1.6° (0.7) | 215 (83.01) | |
| IFN‐beta 1a 44 μg | 386 | 69 | 36.7 (9.8) | 2.35 (1.28) | 5.93 (6.25) | 0.97** (0.42) | 150 (39) | |
| GA | 378 | 72 | 36.8 (9.5) | 2.33 (1.31) | 6.55 (7.10) | 1.01** (0.35) | 156 (41) | |
| IFN‐beta 1b | 897 | 70 | 35.8** (11.13) | 2.35 (2) | 5.3** (4.45) | 1.6° (0.74) | ND | |
| GA | 448 | 68 | 35.2** (11.87) | 2.28 (2) | 5.1** (4.45) | 1.6° (0.74) | ND | |
| ND (no data available). *Median (range); mean not reported. **SD or weighted mean was calculated. °Pre‐1‐year. | ||||||||
| Study | Arm | N° participants | N° dropout | Reasons for missing | |||||||||||
| At randomisation | 24 months | 36 months | n° | % | Lost to follow‐up | Did not receive drug | Switched treatment | Withdrew | Deviated from protocol | Had adverse events | Died | Became pregnant | Other reasons | ||
| IFN‐beta 1a 44 μg | 36 | 25 | ND | 116 | 30.6 | 7 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 38 | |
| GA | 39 | 31 | ND | 87 | 20.5 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 48 | |
| IFN‐beta 1a 44 μg | 55 | 46 | ND | 9 | 16.4 | 9 | ND | ||||||||
| IFN‐beta 1a 30 μg | 55 | 47 | ND | 8 | 14.5 | 8 | |||||||||
| GA | 55 | 48 | ND | 7 | 12.7 | 7 | |||||||||
| IFN‐beta 1a 30 μg | 250 | ND | 194 | 56 | 22.4 | 13 | 0 | 0 | 0 | 0 | 1711 | 1 | 0 | 2513 | |
| GA | 259 | ND | 223 | 36 | 13.9 | 9 | 0 | 0 | 0 | 0 | 1112 | 1 | 0 | 1514 | |
| IFN‐beta 1a 44 μg | 386 | 301 | ND | 85 | 22.0 | 17 | 3 | 0 | 0 | 2 | 23 | 0 | 8 | 329 | |
| GA | 378 | 324 | ND | 54 | 14.3 | 2 | 3 | 0 | 0 | 2 | 19 | 0 | 5 | 2310 | |
| IFN‐beta 1b 250 | 897 | 784 | ND | 113 | 12.6 | 12 | 9 | 10 | 38 | 3 | 13 | 1 | 27 | ||
| GA | 448 | 374 | ND | 74 | 16.5 | 12 | 3 | 5 | 18 | 2 | 8 | 1 | 1 | 24 | |
| ND (no data available) Source of data are described as follow. 1] pg 1977; 2] pg 3; 3] pg 33; 4] pg 904; 5] pg 890; 6] 7 lost to follow‐up + 4 discontinued intervention (see pg 1977); 7] 4 lost to follow‐up + 4 discontinued intervention (see pg 1977); 8] treatment failure; 9] 4 had disease progression + 28 for other reasons; 10] 7 had disease progression + 16 for other reasons; 11] 4 AE/SAE + 13 side effects; 12] 6 AE/SAE + 5 side effects; 13] 14 non‐medical reason + 11 other medical reason; 14] 8 non‐medical reason + 7 other medical reason | |||||||||||||||
| Study | Arm | N° participants | N° dropout | Lost to follow‐up | |||
| Randomised available for the outcome | 24 months | 36 months | n° | % | |||
| IFN‐beta 1a 44 | 36 | 25 | 11 | 30.6 | 7 | ||
| GA | 39 | 31 | 8 | 20.5 | 4 | ||
| IFN‐beta 1a 44 | 55 | 46 | 9 | 16.4 | 9 | ||
| IFN‐beta 1a 30 | 55 | 47 | 8 | 14.5 | 8 | ||
| GA | 55 | 48 | 7 | 12.7 | 7 | ||
| IFN‐beta 1a 30 | 250 | 194 | 56 | 22.4 | 13 | ||
| GA | 259 | 223 | 36 | 13.9 | 9 | ||
| IFN‐beta 1a 44 | 386 | 301 | 85 | 22.0 | 17 | ||
| GA | 378 | 324 | 54 | 14.3 | 2 | ||
| IFN‐beta 1b | 897 | 784 | 113 | 12.6 | 57* | ||
| GA | 448 | 374 | 74 | 16.5 | 31* | ||
| *Data provided by Bayer (Pleimes 2013). | |||||||
| Study | Arm | N° participants at | N° dropout | Lost to follow‐up | |||
| Randomised available for the outcome | 24 months | 36 months | n° | % | |||
| IFN‐beta 1a 44 | 36 | 25 | 11 | 30.6 | 7 | ||
| GA | 39 | 31 | 8 | 20.5 | 4 | ||
| IFN‐beta 1a 44 | 55 | 46 | 9 | 16.4 | 9 | ||
| IFN‐beta 1a 30 | 55 | 47 | 8 | 14.5 | 8 | ||
| GA | 55 | 48 | 7 | 12.7 | 7 | ||
| IFN‐beta 1a 30 | 241 | 194 | 47 | 19.5 | 13 | ||
| GA | 246 | 223 | 23 | 9.3 | 9 | ||
| IFN‐beta 1a 44 | 386 | 301 | 85 | 22.0 | 17 | ||
| GA | 378 | 324 | 54 | 14.3 | 2 | ||
| IFN‐beta 1b | 886 | 784 | 102 | 11.5 | 80* | ||
| GA | 444 | 374 | 70 | 15.8 | 58* | ||
| *Data provided by Bayer (Pleimes 2013). | |||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 At the end of follow‐up (24 ‐ 36 months) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 At 24 months | 3 | 2184 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.87, 1.24] |
| 1.2 At 36 months | 1 | 509 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.92, 1.75] |
| 2 During follow‐up likely scenario Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 At 24 months | 3 | 2184 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.91, 1.34] |
| 2.2 At 36 months | 1 | 509 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.98, 1.73] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 At the end of follow‐up (24‐36 months) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 At 24 months | 3 | 2169 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.91, 1.35] |
| 1.2 At 36 months | 1 | 487 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.63, 1.20] |
| 2 During follow‐up likely scenario Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 At 24 months | 3 | 2169 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.81, 1.90] |
| 2.2 At 36 months | 1 | 487 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.71, 1.26] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Number of participants who dropped out for AE Show forest plot | 4 | 2685 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.64, 1.40] |
| 2 Number of participants who dropped out for SAE Show forest plot | 3 | 2610 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.63, 1.56] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Relapse frequency Show forest plot | 5 | Rate Ratio (Random, 95% CI) | Subtotals only | |
| 1.1 At 24 months | 4 | Rate Ratio (Random, 95% CI) | 1.06 [0.95, 1.18] | |
| 1.2 At 36 months | 1 | Rate Ratio (Random, 95% CI) | 1.40 [1.13, 1.74] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Time to first relapse (HR) Show forest plot | 3 | Hazard Ratio (Random, 95% CI) | 1.01 [0.87, 1.16] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Patients Treated with Steroids Show forest plot | 2 | 1420 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.76, 2.24] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 During follow‐up Show forest plot | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 At 6 months | 1 | 396 | Mean Difference (IV, Random, 95% CI) | ‐0.86 [‐1.32, ‐0.40] |
| 1.2 At 12 months | 2 | 1722 | Mean Difference (IV, Random, 95% CI) | ‐0.52 [‐1.12, 0.09] |
| 1.3 At 24 months | 3 | 1790 | Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.68, 0.39] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean number Show forest plot | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 At 6 months | 0 | 0 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 1.2 At 12 months | 1 | 1233 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.26, 0.06] |
| 1.3 At 24 months | 3 | 1734 | Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.30, 0.02] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean absolute change Show forest plot | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 At 12 months | 1 | 1221 | Mean Difference (IV, Random, 95% CI) | ‐0.4 [‐0.59, ‐0.21] |
| 1.2 At 24 months | 2 | 1608 | Mean Difference (IV, Random, 95% CI) | ‐0.58 [‐0.99, ‐0.18] |
| 1.3 At 36 months | 1 | 509 | Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐1.04, 0.52] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean absolute change Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 At 12 months | 1 | 1207 | Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.18, 0.07] |
| 1.2 At 24 months | 2 | 1602 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.33, ‐0.07] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean absolute change Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 At 12 months | 1 | 1137 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.22, 0.02] |
| 1.2 At 24 months | 2 | 1552 | Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.23, ‐0.01] |