Krótkoterminowe i odległe efekty stosowania tybolonu u kobiet w okresie pomenopauzalnym
Abstract
Background
Tibolone is a synthetic steroid used for the treatment of menopausal symptoms, on the basis of short‐term data suggesting its efficacy. We considered the balance between the benefits and risks of tibolone.
Objectives
To evaluate the effectiveness and safety of tibolone for treatment of postmenopausal and perimenopausal women.
Search methods
In October 2015, we searched the Gynaecology and Fertility Group (CGF) Specialised Register, the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, Embase and PsycINFO (from inception), the Cumulative Index to Nursing and Allied Health Literature (CINAHL) and clinicaltrials.gov. We checked the reference lists in articles retrieved.
Selection criteria
We included randomised controlled trials (RCTs) comparing tibolone versus placebo, oestrogens and/or combined hormone therapy (HT) in postmenopausal and perimenopausal women.
Data collection and analysis
We used standard methodological procedures of The Cochrane Collaboration. Primary outcomes were vasomotor symptoms, unscheduled vaginal bleeding and long‐term adverse events. We evaluated safety outcomes and bleeding in studies including women either with or without menopausal symptoms.
Main results
We included 46 RCTs (19,976 women). Most RCTs evaluated tibolone for treating menopausal vasomotor symptoms. Some had other objectives, such as assessment of bleeding patterns, endometrial safety, bone health, sexuality and safety in women with a history of breast cancer. Two included women with uterine leiomyoma or lupus erythematosus.
Tibolone versus placebo
Vasomotor symptoms
Tibolone was more effective than placebo (standard mean difference (SMD) ‐0.99, 95% confidence interval (CI) ‐1.10 to ‐0.89; seven RCTs; 1657 women; moderate‐quality evidence), but removing trials at high risk of attrition bias attenuated this effect (SMD ‐0.61, 95% CI ‐0.73 to ‐0.49; odds ratio (OR) 0.33, 85% CI 0.27 to 0.41). This suggests that if 67% of women taking placebo experience vasomotor symptoms, between 35% and 45% of women taking tibolone will do so.
Unscheduled bleeding
Tibolone was associated with greater likelihood of bleeding (OR 2.79, 95% CI 2.10 to 3.70; nine RCTs; 7814 women; I2 = 43%; moderate‐quality evidence). This suggests that if 18% of women taking placebo experience unscheduled bleeding, between 31% and 44% of women taking tibolone will do so.
Long‐term adverse events
Most of the studies reporting these outcomes provided follow‐up of two to three years (range three months to three years).
Breast cancer
We found no evidence of differences between groups among women with no history of breast cancer (OR 0.52, 95% CI 0.21 to 1.25; four RCTs; 5500 women; I2= 17%; very low‐quality evidence). Among women with a history of breast cancer, tibolone was associated with increased risk (OR 1.5, 95% CI 1.21 to 1.85; two RCTs; 3165 women; moderate‐quality evidence).
Cerebrovascular events
We found no conclusive evidence of differences between groups in cerebrovascular events (OR 1.74, 95% CI 0.99 to 3.04; four RCTs; 7930 women; I2 = 0%; very low‐quality evidence). We obtained most data from a single RCT (n = 4506) of osteoporotic women aged 60 to 85 years, which was stopped prematurely for increased risk of stroke.
Other outcomes
Evidence on other outcomes was of low or very low quality, with no clear evidence of any differences between the groups. Effect estimates were as follows:
• Endometrial cancer: OR 2.04, 95% CI 0.79 to 5.24; nine RCTs; 8504 women; I2 = 0%.
• Cardiovascular events: OR 1.38, 95% CI 0.84 to 2.27; four RCTs; 8401 women; I2 = 0%.
• Venous thromboembolic events: OR 0.85, 95% CI 0.37 to 1.97; 9176 women; I2 = 0%.
• Mortality from any cause: OR 1.06, 95% CI 0.79 to 1.41; four RCTs; 8242 women; I2 = 0%.
Tibolone versus combined HT
Vasomotor symptoms
Combined HT was more effective than tibolone (SMD 0.17, 95% CI 0.06 to 0.28; OR 1.36, 95% CI 1.11 to 1.66; nine studies; 1336 women; moderate‐quality evidence). This result was robust to a sensitivity analysis that excluded trials with high risk of attrition bias, suggesting a slightly greater disadvantage of tibolone (SMD 0.25, 95% CI 0.09 to 0.41; OR 1.57, 95% CI 1.18 to 2.10). This suggests that if 7% of women taking combined HT experience vasomotor symptoms, between 8% and 14% of women taking tibolone will do so.
Unscheduled bleeding
Tibolone was associated with a lower rate of bleeding (OR 0.32, 95% CI 0.24 to 0.41; 16 RCTs; 6438 women; I2 = 72%; moderate‐quality evidence). This suggests that if 47% of women taking combined HT experience unscheduled bleeding, between 18% and 27% of women taking tibolone will do so.
Long‐term adverse events
Most studies reporting these outcomes provided follow‐up of two to three years (range three months to three years). Evidence was of very low quality, with no clear evidence of any differences between the groups. Effect estimates were as follows:
• Endometrial cancer: OR 1.47, 95% CI 0.23 to 9.33; five RCTs; 3689 women; I2 = 0%.
• Breast cancer: OR 1.69, 95% CI 0.78 to 3.67; five RCTs; 4835 women; I2 = 0%.
• Venous thromboembolic events: OR 0.44, 95% CI 0.09 to 2.14; four RCTs; 4529 women; I2 = 0%.
• Cardiovascular events: OR 0.63, 95% CI 0.24 to 1.66; two RCTs; 3794 women; I2 = 0%.
• Cerebrovascular events: OR 0.76, 95% CI 0.16 to 3.66; four RCTs; 4562 women; I2 = 0%.
• Mortality from any cause: only one event reported (two RCTs; 970 women).
Authors' conclusions
Moderate‐quality evidence suggests that tibolone is more effective than placebo but less effective than HT in reducing menopausal vasomotor symptoms, and that tibolone is associated with a higher rate of unscheduled bleeding than placebo but with a lower rate than HT.
Compared with placebo, tibolone increases recurrent breast cancer rates in women with a history of breast cancer, and may increase stroke rates in women over 60 years of age. No evidence indicates that tibolone increases the risk of other long‐term adverse events, or that it differs from HT with respect to long‐term safety.
Much of the evidence was of low or very low quality. Limitations included high risk of bias and imprecision. Most studies were financed by drug manufacturers or failed to disclose their funding source.
PICOs
Streszczenie prostym językiem
Krótkoterminowe i odległe efekty stosowania tybolonu u kobiet w okresie pomenopauzalnym
Pytanie badawcze
Celem autorów przeglądu Cochrane była ocena skuteczności i bezpieczeństwa stosowania tybolonu w leczeniu kobiet w okresie pomenopauzalnym i okołomenopauzalnym.
Wprowadzenie
Tybolon stanowi dostępną opcję leczenia objawów menopauzy, a dane krótkoterminowe wskazują na jego skuteczność. Jednak świadczeniodawcy muszą uwzględniać równowagę pomiędzy korzyściami i zagrożeniami wynikającymi ze stosowania tybolonu, gdyż pojawiły się obawy, że jego stosowanie może być związane z występowaniem udaru mózgu, raka piersi oraz raka endometrium.
Charakterystyka badania
Włączyliśmy 46 badań z randomizacją (RCTs), które obejmowały łącznie 19 976 kobiet w okresie pomenopauzalnym. W większości badań oceniano zastosowanie tybolonu w leczeniu objawów naczynioruchowych w okresie menopauzy. W niektórych badaniach zgłaszano również inne cele: celem czterech badań z randomizacją (badania z losowym przydziałem uczestników do grupy badanej i kontrolnej ‐ przyp. tłum.) była ocena bezpieczeństwa odnośnie endometrium, czterech ‐ profil krwawień, pięciu ‐ zapobieganie utracie masy kostnej i złamaniom kości, jednego ‐ ocena sfery seksualnej oraz trzech ‐ ocena bezpieczeństwa u kobiet z rakiem piersi w wywiadach, w dwóch badaniach oceniano stosowanie tybolonu u kobiet z mięśniakami lub toczniem rumieniowatym. Dane są aktualne do października 2015 r.
Główne wyniki
Dane naukowe o średniej jakości wskazują, że tybolon jest bardziej skuteczny niż placebo ale mniej skuteczny niż skojarzona terapia hormonalna w zmniejszaniu nasilenia objawów naczynioworuchowych u kobiet po menopauzie. Dane te sugerują również, że jeśli 67% kobiet przyjmujących placebo doświadczało objawów naczynioruchowych, to 35% ‐ 45% kobiet przyjmujących tybolon również ich doświadczy, a także, że jeżeli 7% kobiet przyjmujących skojarzoną terapię hormonalną doświadczyło objawów naczynioruchowych, to u 8% ‐ 14% kobiet przyjmujących tybolon również wystąpią wspomniane objawy. Ponadto, dane naukowe o średniej jakości wskazują, że stosowanie tybolonu wiąże się z częstszym występowaniem nieplanowanych krwawień w porównaniu z placebo, jednak w mniejszym stopniu niż w czasie stosowania terapii hormonalnej.
W porównaniu z placebo, tybolon zwiększa ryzyko nawrotu raka piersi u kobiet, które w przeszłości chorowały na ten nowotwór oraz może zwiększać ryzyko wystąpienia udaru mózgu u kobiet powyżej 60 roku życia. Nie odnaleziono wyników badań sugerujących, że tybolon zwiększa ryzyko innych poważnych zdarzeń niepożądanych, nie ma też danych ukazujących różnice pomiędzy tybolonem i terapią hormonalną w odniesieniu do odległych działań niepożądanych. Prawie wszystkie dane naukowe dotyczące działań niepożądanych były bardzo niskiej jakości, a zgłaszane zdarzenia były rzadkie.
Jakość danych naukowych
Znaczna część uzyskanych danych naukowych była niskiej lub bardzo niskiej jakości. Ograniczenia obejmowały duże ryzyko wystąpienia błędu systematycznego (wypaczenie wyników badania ‐ przyp. tłum.) we włączonych badaniach, bardzo małą liczbę zdarzeń w stosunku do liczby uczestników oraz potencjalne konflikty interesów. Spośród włączonych badań dwadzieścia sześć było finansowanych przez producentów leków, a w kolejnych 14 badaniach nie ujawniono źródła finansowania.
Authors' conclusions
Summary of findings
| Tibolone compared with placebo: vasomotor symptoms | ||||||
| Population: postmenopausal women with vasomotor symptoms | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Tibolone | |||||
| Vasomotor symptoms (all doses) Follow‐up: 12 weeks to 1 year | 670 per 1000 | 400 per 1000 (350 to 450) | OR 0.33 | 842 (5 RCTs) | ⊕⊕⊝⊝ | Three studies at high risk of attrition bias were excluded from this analysis. Inclusion of these studies was associated with stronger effect of tibolone but with extreme heterogeneity (I2= 97%) |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level for serious risk of bias: poor reporting of study methods and potential conflict of interest (pharmaceutical funding) in most studies; standard deviations imputed for some studies. Effect estimate robust to a sensitivity analysis excluding studies at high risk of attrition bias | ||||||
| Tibolone compared with placebo: adverse events | ||||||
| Population: postmenopausal women with or without vasomotor symptoms | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Tibolone | |||||
| Endometrial cancer (all doses) | See comment | OR 2.04 | 8504 | ⊕⊝⊝⊝ | Events very rare in both groups. Total of 21 events: 16/4486 in tibolone group, 5/4018 in placebo group | |
| Breast cancer; women without previous breast cancer (all doses) | 4 per 1000 | 1 per 1000 (1 to 5) | OR 0.52 | 5500 | ⊕⊝⊝⊝ | In women with a history of breast cancer, risk increased in the tibolone group at 1 to 2.75 years' follow up: OR 1.50 (1.21 to 1.85, 2 RCTs, 3165 women, moderate‐quality evidence ) |
| Unscheduled bleeding (all doses) Follow‐up: 1 to 3 years (median 2) | 177 per 1000 | 374 per 1000 (310 to 442) | OR 2.79 | 7814 | ⊕⊕⊝⊝ | |
| Venous thromboembolic events (clinical evaluation) all doses | See comment | OR 0.85 | 9176 | ⊕⊝⊝⊝ | Events very rare in both groups. Total of 24 events: 12/5054 in tibolone group, 12/4122 in placebo group | |
| Cardiovascular events (all doses) Follow‐up: 2 to 3 years (median 2.75) | 10 per 1000 | 13 per 1000 (8 to 22) | 1.38 (0.84 to 2.27) | 8401 (4 studies) | ⊕⊝⊝⊝ | |
| Cerebrovascular events (all doses) | 5 per 1000 | 8 per 1000 (4 to 14) | OR 1.74 | 7930 | ⊕⊝⊝⊝ | |
| Mortality from any cause (all doses) | 10 per 1000 | 10 per 1000 (8 to 14) | OR 1.06 | 8242 | ⊕⊕⊝⊝ | |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded two levels for very serious risk of bias: poor reporting of study methods, high attrition and/or potential conflict of interest in most studies bDowngraded one level for serious imprecision: low event rate. Findings compatible with meaningful benefit in one or both arms, or with no effect cDowngraded one level for serious risk of low applicability: Some studies compare doses of tibolone that have not been marketed (although downgrading has no effect on rating, as study already rated very low) dDowngraded one level for serious risk of bias: poor reporting of study methods and potential conflict of interest in most studies eDowngraded one level for potential conflict of interest (funding by pharmaceutical companies) | ||||||
| Tibolone compared with combined HT for postmenopausal women: vasomotor symptoms | ||||||
| Population: postmenopausal women with vasomotor symptoms | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Combined HT | Tibolone | |||||
| Vasomotor symptoms (tibolone 2.5 mg/d) | 70 per 1000 | 110 per 1000 (80 to 140) | OR 1.57 | 646 (4 studies) | ⊕⊝⊝⊝ | From a sensitivity analysis excluding studies with high risk of attrition bias. An inclusive analysis (9 studies, 1336 participants) suggests a similar but slightly reduced disadvantage of tibolone (OR (95% CI) 1.36 (1.11 to 1.66)) |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level for serious risk of bias: poor reporting of study methods and potential conflict of interest in all studies. Effect estimate robust to a sensitivity analysis excluding studies at high risk of attrition bias | ||||||
| Tibolone compared with combined HT for postmenopausal women: adverse events | ||||||
| Population: postmenopausal women with or without vasomotor symptoms | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Combined HT | Tibolone | |||||
| Unscheduled bleeding (all doses) | 474 per 1000 | 224 per 1000 | OR 0.32 | 6438 | ⊕⊕⊝⊝ | |
| Endometrial cancer (all doses) | See comments | OR 1.47 | 3689 | ⊕⊝⊝⊝ | Events very rare in both groups. Total of 3 events: 2/1826 in tibolone group, 1/1863 in combined HT group | |
| Breast cancer; women without previous breast cancer (all doses) | 3 per 1000 | 6 per 1000 (3 to 13) | OR 1.69 | 4835 | ⊕⊝⊝⊝ | |
| Venous thromboembolic events (clinical evaluation; all doses) | 3 per 1000 | 1 per 1000 (0 to 6) | OR 0.44 | 4529 | ⊕⊝⊝⊝ | |
| Cardiovascular events (all doses) | 17 per 1000 | 10 per 1000 (4 to 27) | OR 0.63 | 3794 | ⊕⊝⊝⊝ | |
| Cerebrovascular event (all doses) | 1 per 1000 | 1 per 1000 (0 to 3) | OR 0.76 | 4562 | ⊕⊝⊝⊝ | |
| Mortality from any cause (tibolone 2.5 mg/d) | See comments | OR 3.05 | 970 | ⊕⊝⊝⊝ | Only 1 event (in tibolone group): 1/485 vs 0/485 | |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level for serious risk of bias: poor reporting of study methods and potential conflict of interest in some studies bDowngraded two levels for very serious risk of bias: poor reporting of study methods and potential conflict of interest in some studies cDowngraded one level for serious imprecision: low event rate. Findings compatible with meaningful benefit in one or both arms, or with no effect | ||||||
Background
Description of the condition
Hot flushes are among the most characteristic clinical symptoms of menopause (Politi 2008); they are probably caused by lability in the hypothalamic thermoregulatory centre induced by reduction of oestrogen and progesterone levels (Freedman 1995). Hot flushes and sweats of increasing severity can occur during the night, leading to sleep problems (Porter 1996). Hot flushes and sweats are described as vasomotor symptoms.
Many postmenopausal women report a variety of symptoms such as vaginal dryness (Suckling 2006), sexual discomfort, urinary incontinence (Cody 2012) and frequent urinary infection, probably resulting from the natural decline of oestrogen levels (Speroff 2004).
All symptoms tend to fluctuate, and their perceived severity varies greatly among individuals, with some reporting intense discomfort and a substantial reduction in quality of life.
Researchers have successfully used oestrogens and progestogens to ameliorate vasomotor (MacLennan 2004) and vaginal symptoms (Suckling 2006), anxiety and low mood (NCC‐WCH 2015). Urinary tract infections are less clearly influenced by combined hormone therapy (HT) (Soc Obstetr Gynaecol Canada 2014).
Description of the intervention
Tibolone (Livial®, ORG OD 14) is a synthetic steroid widely prescribed to postmenopausal women in Europe.
How the intervention might work
After its commercialisation, tibolone gained some popularity for combining oestrogenic and progestogen actions. Its mechanism of action is not well known, although many studies, most sponsored by the drug manufacturer, indicate that the drug undergoes different tissue‐selective metabolic transformations and may exert weak oestrogen, progestogen and/or androgen activities (Modelska 2002). The oestrogenic effects, exerted mainly in brain, bone and vaginal tissues, are weaker on the endometrium, where the drug is transformed into progestogen metabolites. In breast tissue, limited conversion of oestrone to oestradiol may reduce the oestrogenic effects. In brain and liver, tibolone seems to have androgenic effects. Some randomised controlled clinical trials (RCTs) have suggested that tibolone decreases vasomotor symptoms and ameliorates vaginal dryness and discomfort, but results are not consistent. An RCT published in 2009 (Kenemans 2009) highlighted that tibolone increases recurrence of breast cancer, revealing a contraindication for women with a history of breast cancer. Although the drug is thought to have a possible role in preserving bone mineral density, control of osteoporosis is not a recommended indication.
Why it is important to do this review
The safety profile of tibolone has not been well defined, and trials evaluating its use to treat patients with vasomotor symptoms usually provide follow‐up periods that are too short for assessment of potential long‐term adverse events such as increased risk of endometrial (Beral 2005) and breast (Kenemans 2009; Beral 2003) cancer and of cardiovascular events (Cummings 2008). For this reason, safety has been evaluated in a wider population, and RCTs including women who did not take tibolone for symptomatic relief have been considered.
Objectives
To evaluate the effectiveness and safety of tibolone for treatment of postmenopausal and perimenopausal women.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs). We did not include quasi‐randomised and cross‐over trials.
Types of participants
Menopausal and perimenopausal women with or without vasomotor and/or genital symptoms, defined as women with surgical menopause or with spontaneous menopause, or women who had menstruated irregularly over the past 12 months.
Types of interventions
-
Tibolone use versus placebo
-
Tibolone use versus oestrogens
-
Tibolone use versus combined HT (referring to two different formulations: sequential combined and continuous combined)
This review did not consider tibolone use versus no treatment.
Types of outcome measures
Primary outcomes
-
Vasomotor symptoms measured as occurrences or through scales, defined as any otherwise unexplained sensation of flushing/sweating experienced by the participant. We included studies that measured hot flushes (with or without night sweats), provided that they measured hot flushes as an outcome of efficacy in populations including symptomatic women
-
Unscheduled bleeding (vaginal bleeding and/or spotting)
-
Long‐term adverse events: endometrial cancer, breast cancer, venous thromboembolic events, cardiovascular events, cerebrovascular events, mortality from any cause
Secondary outcomes
-
Insomnia (frequency or continuous outcome)
-
Genital symptoms: vaginal dryness and painful sexual intercourse (measured as frequency or severity), vaginal infection (inflammation of the vagina usually related to one of three infectious conditions: bacterial vaginosis, vulvovaginal candidiasis, trichomoniasis), urinary tract infection
-
Endometrial hyperplasia
We measured all outcomes other than vasomotor symptoms in women with or without vasomotor symptoms.
We included studies assessing at least one of these specific outcomes, even if they did not report useable data. We excluded studies not assessing such outcomes.
Search methods for identification of studies
Electronic searches
We searched for all relevant published and unpublished RCTs, without language restriction, and in consultation with the Cochrane Gynaecology and Fertility Group (CGF) Information Specialist.
We searched the CGF Specialised Register (formerly known as the Menstrual Disorders and Subfertility Group Specialised Register), the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, Embase, PsycINFO and the Cumulative Index to Nursing and Allied Health Literature (CINAHL), from inception until 15 October 2015, using the strategies shown in Appendix 1, Appendix 2, Appendix 3, Appendix 4, Appendix 5 and Appendix 6. For the search of clinicaltrials.gov, we used "tibolone" as a keyword. We contacted individual researchers and the current manufacturer of tibolone to ask them to identify unpublished and ongoing trials.
Searching other resources
We contacted individual researchers working in relevant fields (gynaecology, endocrinology) and the current manufacturer of tibolone (Merck Sharp & Dome) to check for additional relevant references and unpublished and ongoing trials. We also checked the reference lists of all studies identified by the above methods.
Data collection and analysis
Selection of studies
Four review authors (GF, EP, SM, SB) independently screened the titles and abstracts of articles found in the search for inclusion. We searched for outcomes of interest in the full texts, even if they had not been reported in the abstracts. We resolved disagreements by discussion and by consultation with two additional review authors (VB, a gynaecologist; and EM, an endocrinologist). We sought further information from study authors who published papers containing insufficient information to permit a decision about eligibility. We recorded reasons for excluding studies after debate and agreement.
Data extraction and management
Five review authors (GF, EP, SM, SB, JW) independently extracted details of study design, participants, interventions, follow‐up, quality components, efficacy outcomes and adverse events.
Three other review authors (VB, a gynaecologist; EM, an endocrinologist; and AMM, a cardiologist) resolved discrepancies regarding extraction of quantitative data or risk of bias assessment of RCTs. When a trial was presented in abstract form, we sought further information by searching the Internet, by contacting study authors and by checking for the next best available resource or publication. We contacted study authors for further insight on study design and results, when we considered this necessary. For studies with more than one publication, we extracted data from all publications, but we considered the final or updated version of each trial to be the primary reference.
We extracted the following information from the studies included in the review (see also Characteristics of included studies table).
Trial characteristics
-
Randomisation
-
Allocation concealment
-
Trial design: multi‐centre or single‐centre
-
Number of women randomised, excluded and analysed
-
Duration, timing and location of the trial
-
Source of funding and conflicts of interest
Baseline characteristics of studied groups
-
Definition and duration of preexisting menopausal condition
-
Age of the women
-
Previously administered treatment(s)
Interventions
-
Type of intervention and control
-
Dose regimen
-
Treatment duration
Outcomes
-
Outcomes reported
-
Definitions of outcomes
-
The way outcomes were measured
-
Timing of outcome measurement
If data were reported only in figures, we used Microsoft PowerPoint to extract data from the figures. We opened the figure in the software and overlaid a grid. We drew horizontal or vertical lines as needed, and we ‘snapped’ (aligned) them to this grid, to ensure that they were parallel/perpendicular to the plot axes, as required. We could move lines drawn in the software vertically and horizontally, so we could read off the value corresponding to a given data point in a scatterplot or the height of a bar in a bar chart against the appropriate axis. A single review author (JW) extracted data from figures.
Assessment of risk of bias in included studies
We assessed risk of bias of included trials by taking six components into account: generation of the allocation sequence (participant randomisation), allocation concealment, blinding (or masking) of participants and personnel, blinding of outcome assessment, completeness of follow‐up (attrition bias) and selective reporting. We used the following definitions when assessing risk of bias.
Generation of the allocation sequence
-
Adequate: if the allocation sequence was generated by a computer or by a random number table. We considered drawing of lots, tossing of a coin, shuffling of cards or throwing of die as adequate if a person not otherwise involved in recruitment of participants performed the procedure
-
Unclear: if the trial was described as randomised, but the method used for generation of the allocation sequence was not described
-
Inadequate: if a system involving dates, names or admittance numbers was used for allocation of women. We excluded these studies, known as quasi‐randomised, from the present review
We also excluded trials with alternating allocation.
Allocation concealment
-
Adequate: if allocation of women involved a central, independent unit; an on‐site locked computer; identical appearing numbered drug bottles or containers prepared by an independent pharmacist or investigator; or sealed, opaque envelopes
-
Unclear: if the trial was described as randomised but the method used to conceal the allocation was not described
-
Inadequate: if the allocation sequence was known to investigators who assigned participants, envelopes were unsealed or transparent or the study was quasi‐randomised
Blinding (or masking) of participants and personnel
-
Adequate: if the trial was described as double‐blind and the method of blinding involved identical placebo or active drugs, particularly:
-
double‐blind (method described and use of a placebo(s) or dummy technique meant neither the participant nor the care provider or assessor knew which treatment was given)
-
single‐blind (participant, care provider or assessor was aware of the treatment given)
-
-
Unclear: if the trial was described as double‐blind or single‐blind but the method of blinding was not described
-
Not performed: if the trial was open‐label (all parties aware of treatment)
Blinding of outcome assessment
-
Adequate: if in the absence of blinding of outcome assessment, review authors judged that outcome measurement was not likely to be influenced by lack of blinding; or if blinding of outcome assessment was ensured and it was unlikely that blinding could have been broken
-
Unclear: if information was insufficient to permit judgement of ‘low risk’ or ‘high risk’, or if the study did not address this outcome
-
Inadequate: if no blinding of outcome assessment occurred and outcome measurement was likely to be influenced by lack of blinding; or if blinding of outcome assessment was present but blinding could have been broken, and if outcome measurement was likely to be influenced by lack of blinding
Completeness of follow‐up (attrition bias)
-
Adequate: if numbers and reasons for dropouts and withdrawals in all intervention groups were described and 90% or more of randomised participants were included in the analysis; or if it was specified that no dropouts or withdrawals occurred
-
Unclear: if the report gave the impression that no dropouts or withdrawals occurred but this was not specifically stated
-
Inadequate: if less than 90% of randomised participants were included in the analysis; or numbers or reasons for dropouts and withdrawals were not provided
We contacted the authors of primary trial reports when necessary to request clarification of data and to obtain missing information.
Selective reporting
-
Adequate: if the study protocol was available and all of the study’s prespecified (primary and secondary) outcomes of interest in the review were reported in the prespecified way
-
Unclear: if information was insufficient to permit judgement of ‘low risk’ or ‘high risk’
-
Inadequate: if not all of the study’s prespecified primary outcomes were reported; if one or more primary outcomes were reported via measurements, analysis methods or subsets of data (e.g. subscales) that were not prespecified; if one or more reported primary outcomes were not prespecified (unless clear justification for their reporting was provided, such as an unexpected adverse effect); if one or more outcomes of interest in the review were reported incompletely and could not be included in a meta‐analysis; or if the study report failed to include results for a key outcome that would have been expected to be reported for such a study
Measures of treatment effect
We evaluated efficacy and safety outcomes by considering the number of women in the control and intervention groups of each study experiencing at least one event (dichotomous outcomes) to calculate Mantel‐Haenszel odds ratios (DerSimonian 1986) with 95% confidence intervals (CIs), or (for continuous outcomes) mean scores, standard deviations and the number of women in each group, using the inverse variance method. The primary outcome ‘vasomotor symptoms’ and the secondary outcomes vaginal dryness and sleep were exceptions; we reported these outcomes as binary or continuous variables ‐ the first two using several scales. Accordingly, we converted all treatment effect estimates from binary or continuous variables to standardised mean differences (SMDs), as this permitted pooling of these variants in a meta‐analysis. Pooled SMDs computed in this manner can be transformed and interpreted as odds ratios, at the cost of information related to symptom severity (Higgins 2011).
Unit of analysis issues
This systematic review considered only RCTs. The unit of analysis in each RCT was the women who were randomised to one of the treatment arms. For vaginal bleeding, we considered endometrial hyperplasia and endometrial cancer only in women with a uterus.
Dealing with missing data
We analysed data on an intention‐to‐treat basis as far as possible by including all randomised participants in the groups to which they were allocated. Missing data in included studies compromised realisation of this strategy. Moreover, options to rectify the matter were limited in the absence of individual participant data. Accordingly, we took the approach of penalising trials with notable rates of attrition in the risk of bias assessment and conducting sensitivity analyses that were restricted to trials with low risk of bias in this domain. We incorporated these sensitivity analyses into our conclusions.
Assessment of heterogeneity
We included in the meta‐analysis all outcomes reported by individual studies, noting heterogeneity by using Chi2 and I2 statistics (Higgins 2002). We stated that the Chi2 statistic was statistically significant if P < 0.10. The I2statistic indicated the percent of variability due to between‐study (or interstudy) variability, as opposed to within‐study (or intrastudy) variability. We considered an I2value greater than 50% to be large (Higgins 2002). When statistically significant heterogeneity existed, we conducted a careful clinical review of the data to seek the source of such heterogeneity and to decide whether statistical combining of trials was warranted.
Assessment of reporting biases
We graphically assessed publication bias by using contour‐enhanced funnel plots.
Data synthesis
We used a random‐effects model, except for vasomotor symptoms, vaginal dryness and sleep, for which we combined data from dichotomous and continuous outcomes in a fixed‐effect model by converting all treatment effect estimates to standardised mean differences (SMDs). We deemed this necessary because the key assumption of random‐effects meta‐analysis ‐ that all observed treatment effects represent realisations from a common underlying distribution ‐ did not appear to be warranted, given the diversity of outcome reporting scales used. Poor reporting standards required that we impute standard deviations for several studies reporting on menopausal symptoms to combine their results; we calculated all effect sizes and corresponding standard errors by using the metaphor package (Viechtbauer 2010) in R (R Core Team 2015). If results for this outcome were available at several time points, we used results corresponding to the longest period of use. Table 1 and Table 2 provide details of methods used in analyses of menopausal symptoms and vaginal dryness, as well as reasons for exclusion of several RCTs from these meta‐analyses.
| Study | Comparator | Outcome measure | Information available | Notes | Results for meta‐analysis | SMD |
| Al‐Azzawi 1999 | HT | Presence of vasomotor symptoms, severity measured by Greene menopausal symptoms scale | 6 HRT and 9 tibolone patients were without symptoms at baseline. 67 HRT and 58 tibolone patients were free at month 3 | Contacted study authors, no reply | ||
| Baracat 2002 | HT | Total score: mean number of hot flushes per day multiplied by severity score | Means plotted as bar chart in Figure 1. Baseline, 11 for tibolone (n = 40), 12 for control (n = 45). At 3 months, 1.8 for tibolone and 1.5 for control. At 13 months, 0.2 for both | Would have to impute SDs – ‘no significant difference’ Unclear how to do this, given the available info Unable to find contact details | ||
| Benedeck‐Jaszmann 1987 | Placebo | 0 to 3 severity score | 12 months From Fig 1: Mean P: 1.6 T: 0.6 SD P: 1 T: 0.9 N P: 19 T: 24 (assuming 30 per arm to start, not explicitly stated) | Extracted from figure | Mean P: 1.6 T: 0.6 SD P: 1 T: 0.9 N P: 19 T: 24 | SMD: ‐1.0384784 SE: 0.3268612 |
| Bouchard 2012 | Placebo | Severity score | Calculate 12 week values P: 1.59 T: 1.16 Sample sizes of 150 (P) and 164 (T) Wk 12 | Use SD from sample size calc, which is in line with other studies | P: Mean 1.59 SD 0.9 N = 150 T: Mean 1.16 SD: 0.9 N = 164 | SMD: ‐0.4766282 SE: 0.1145686 |
| Egarter 1996 | HT | Severity of hot flushes (modified Kupperman Index) | Baseline mean C: 2.1 T: 2.2 6 months C: 0.4 T: 0.4 ‘N/S’ N = 34 (C) N = 62 (T) | Impute SD ‐ unclear how to Contacted study authors: no reply | ||
| Hammar 2007 | HT | Number of hot flushes | Week 48, baseline mean of both groups 6, follow‐up mean ≤ 1 Baseline SD C: 4.40 T: 4.37 N = 241 (C) N = 222 (T) | Use baseline SDs (these appear reasonable, given Landgren 2002) | C: mean 1, SD 4.40; N = 241 T: mean 1, SD 4.37 N = 222 | SMD: 0.00 SE: 0.09302624 |
| Hudita 2003 | Placebo (3 –arm study) | 5‐point severity scale for hot flushes | Week 24 P: 3 T: 1.25 mg: 0.2 T: 2.5mg: 0.1 N = 34 N = 45 N = 41 P < 0.01 for both compared with placebo | Split control group size between 2 arms Used P value to calculate SD Get implausible answers. Used known value instead (e.g. Hammar 1998) | Mean P: 3 T: 1.25 mg: 0.2 T: 2.5 mg: 0.1 N N = 34/2 = 17 N = 45 N = 41 SD P: 0.63 T: 1.25: 0.87 T: 2.5: 0.87 | 1.25 SMD: ‐3.4009511 SE: 0.4175209 2.5 SMD: ‐3.5375963 SE: 0.4371477 |
| Kokcu 2000 | HT | Occurrence of hot flushes | OR: 4.16 (0.75 to 22.9) | 2/19 have symptoms in C 12/19 have symptoms in T | SMD: 1.6236743 SE: 0.5369759 | |
| Landgren 2002 | Placebo (5‐arm study) | Frequency of hot flushes | Read means and SEs at 12 weeks from Figure 1 Mean P = 5.2 T 0.625 = 5 T 1.25 = 2.1 T 2.5 = 1.8 T 5.0 = 1.6 Standard error P = 0.37 T 0.625 = 0.37 T 1.25 = 0.40 T 2.5 = 0.43 T 5.0 = 0.37 Ns (calculated as all evaluable – dropouts ‐this assumes dropout occurred after 1st measurement at week 4) P = 113 T 0.625 = 129 T 1.25 = 124 T 2.5 = 139 T 5.0 = 136 | Read means and SEs from Figure 1 Calculated SDs using SEs and sample sizes Split placebo group size in 4 113/4 = 28.25 | Mean P = 5.2 T 0.625 = 5 T 1.25 = 2.1 T 2.5 = 1.8 T 5.0 = 1.6 SD P = 3.93 T 0.625 = 4.20 T 1.25 = 4.45 T 2.5 = 5.07 T 5.0 = 4.31 N (calculated as all evaluable – dropouts – this assumes dropout occurred after 1st measurement at week 4) P = 28.25 T 0.625 = 129 T 1.25 = 124 T 2.5 = 139 T 5.0 = 136 | 0.625 SMD: ‐0.04792794 SE: 0.20552850 1.25 SMD: ‐0.7077526 SE: 0.2102005 2.5 SMD: ‐0.6912512 SE: 0.2076033 5.0 SMD: ‐0.8437215 SE: 0.2097448 |
| Mendoza 2002 | HT | Flushes subscore of the Modified Kupperman Index, 0 to 2 score Number (%) reduced | Have number and percentage that improved in terms of vasomotor symptoms after 1 year Have 2 possible control groups – choose the best performing to give a conservative estimate 25/26 reduced in control group 27/29 reduced in T groups | Calculate odds ratio for reduced vasomotor symptoms. Turn this into an SMD for combination (27/2)/(25/1) = 0.54 SE log(OR) = Sqrt(1/27+1/2+1/25+1/1) = 1.26 | OR for improvement: OR = 0.54 SE(log(OR)) = 1.26 (so T worse) | SMD: 0.3734461 SE: 0.7610917 |
| Nappi 2006a | HT | Vasomotor symptoms (0 to 3 severity score) | At 6 months Means from Figure 4 C: 1.75 T: 1.5 P value for treatment term in ANOVA given as ‘P < 0.4’ N = 20 in both groups | Assume ANOVA P value is 0.4 and work out SDs as though this was a t‐test Gives SD of 0.657, assuming same in both groups | SMD: ‐0.3729492 SE: 0.3189649 | |
| Ross 1999 | HT | Greene Climacteric Scale subscore | Nothing usable. Only present 1 of 6 relevant comparisons because it is almost significant. Do not present 3 month score | |||
| Siseles 1995 | HT | Kupperman Index | No information given for vasomotor subscale | Have contacted study authors, no reply | ||
| Swanson 2006 | Placebo (3‐arm study) | Number of hot flushes per day | Median change from baseline at week 12 ‐5.5 P ‐9.7 T 2.5 ‐8.3 T 1.25 P < 0.001 for T 2.5 vs P P < 0.003 for T 125 vs P N P: 133 T 2.5: 125 T 1.25: 133 Actually, mean changes at week 12 and P values given in abstract T 2.5 vs P ‐10.14 vs ‐5.85, P < 0.001 T 1.25 vs P, week12 ‐8.32 P < 0.003 | Use reported values and calculate as for t‐tests. Split placebo group in half. Will have to impute SDs and final scores, as changes cannot be pooled with final scores if SMDs are used. For baseline, take median of values from Hammar 2007 and Landgren 2002 6,6,8,8,8,9,9.7 Mean 7.8. Too low – Figure 2 shows large changes. Say, 10 P: 10 ‐ 5.85 = 4.15 T 2.5: 10 ‐ 10 = 0 T 1.25: 10 ‐ 8.32 = 1.68 SDs too large when calculated from t‐test. Use values from Langren: P: 3.93 T 2.5: 5.07 T 1.25: 4.45 | Mean P: 10 ‐ 5.85 = 4.15 T 2.5: 10 ‐ 10 = 0 T 1.25: 10 ‐ 8.32 = 1.68 SD P: 3.93 T 2.5: 5.07 T 1.25: 4.45 N P: 66 T 2.5: 125 T 1.25: 133 | 1.25 SMD: ‐0.5741771133 SE: 0.1532927 2.5 SMD: ‐0.9661562 SE: 0.1599848 |
| Vieira 2009 | Placebo | Kupperman Index | Only overall Kupperman Index shown | Have contacted study authors, no reply | ||
| Volpe 1986 | Placebo HT | 0 to 9 score, with 0 = absent, 3 = mild, 6 = moderate, 9 = severe Unclear whether intermediate scores are possible | Can extract means for 24 weeks for tibolone arm, placebo arm and each of several HT arms, which have been partially combined, from Figure 1 in the paper | No real way to calculate SD from info in the paper, and the scale is different from those used in other studies (so not reasonable to use one from another study) | ||
| Wender 2004 | Placebo | Kupperman Index | Only overall Kupperman Index shown | Have contacted study authors, no reply |

Study flow diagram.
| Study | Comparator | Outcome measure | Information available | Method used | Results for meta‐analysis | SMD |
| Hudita 2003 | Placebo (3‐arm study) | 0 to 4 scale | From figure Week 24 P: 2.6 T 1.25 mg: 1 T 2.5 mg: 0.9 N = 34/2 = 17 N = 45 N = 41 | Split control group size between 2 arms Use known value from other study for SD Use those from Nappi 2006a SD T: 0.89 HT: 0.89 | Mean P: 2.6 T 1.25 mg: 1 T 2.5 mg: 0.9 N N = 34/2 = 17 N = 45 N = 41 SD P: 0.89 T 1.25: 0.89 T 2.5: 0.89 | 1.25mg SMD: ‐1.7751711 SE: 0.3262804 2.5mg SMD: ‐1.8843965 SE: 0.3373802 |
| Kenemans 2009 | Placebo | Vaginal dryness as binary | P: 33/1558 T: 19/1575 | Convert OR to SMD | P: 33/1558 T: 19/1575 | |
| Swanson 2006 | Placebo (3‐arm study) | 0 to 3 score | Mean change from baseline at week 12 P: ‐0.2 T 2.5: ‐0.26 T 1.25: ‐0.39 N P: 133 T 2.5: 125 T 1.25: 133 | Split control group size between 2 arms Calculate final means using baseline and change – but no baseline values given Would also need to use SDs from another study | Cannot use | |
| Huber 2002 | HT | Vaginal dryness as binary | HT: 7/166 T: 6/158 | Convert OR to SMD | HT: 7/166 T: 6/158 | SMD: ‐0.06613757 SE: 0.34411866 |
| Kokcu 2000 | HT | Vaginal dryness as binary | HT: 0/21 T: 1/23 | Convert OR to SMD | HT: 0/21 T: 1/23 | SMD: 0.6382727 SE: 1.0064298 |
| Ziaei 2010 | HT and placebo | Vaginal dryness as binary Also, lubrication scores 1 to 5, higher is better – can reverse signs of mean differences | HT: 20/42 T: 33/47 P: 37/48 Mean HT: 4.93 T: 4.58 P: 3.65 SD HT: 1.95 T: 1.26 P: 1.81 | Use the continuous data Calculate and reverse sign, so that greater = increased vaginal dryness | HT: 20/42 T: 33/47 P: 37/48 | Using OR to SMD vs HT SMD: 0.5774306 0.2691251 vs placebo SMD ‐0.5904427 SE: 0.2096301 Using lubrication scores vs HT: SMD after switching sign: 0.2138954 SE: 0.2129393 vs placebo: SMD after switching sign: ‐0.1313959 SE: 0.2185150 |
| Nappi 2006a | HT | Vaginal dryness 0 to 3 score | From Figure 4, mean at 6 months Mean T: 0.7 HC: 0.6 SD: can read SE off Figure 4 and calculate SD N = 20 both groups | SD: can read SE off Figure 4 and calculate SD T: 0.89 HT: 0.89 | Mean T: 0.7 HC: 0.6 SD T: 0.89 HT: 0.89 N = 20 | SMD: 0.1101248 SE: 0.3164674 |
| Uygur 2005 | HT | 7‐point scale with ‐3 as worsened a lot and 3 as improved a lot | 6 months Mean (higher is better) HT: 0 T: 0.56 N HT: 34 T: 38 | P < 0.05 given. Assume P = 0.05 and calculate SD, assuming equal in 2 groups: Gives SD = 1.7 | Mean (higher is better) HT: 0 T: 0.56 N HT: 34 T: 38 Sd=1.7 for both | SMD after sign change: ‐0.3258676 0.2376236 |
We sought the following comparisons.
-
Tibolone use, stratified by dose, versus placebo.
-
Tibolone use, stratified by dose, versus oestrogens.
-
Tibolone use, stratified by dose, versus combined HT.
To avoid multiple‐counting of a control group in RevMan, we split the numbers of events and of exposed participants in studies with multiple arms, depending on the number of comparisons, as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011; see paragraph 16.5.4). We did not perform this procedure in cases of rare events (e.g. when one or three cases should have been split) or when estimated odds ratios differed by more than 0.05 from the non‐stratified analysis. In the latter case, we combined intervention groups (e.g. different doses of tibolone) to create a single pair‐wise comparison versus the control group.
Subgroup analysis and investigation of heterogeneity
We stratified results according to tibolone dose. Two of the largest RCTs, which assessed the occurrence of breast cancer and cardio‐cerebrovascular events, selected very specific and heterogeneous populations; therefore, we considered that it would be informative to present results on breast cancer separately for women who had a history of breast cancer and those who had no such history, and results on cardiovascular and cerebrovascular events that distinguished women younger than and over 60 years of age. We did not prespecify these subgroup analyses.
Sensitivity analysis
We conducted sensitivity analyses of the primary outcome to determine whether conclusions were robust to arbitrary decisions regarding eligibility and analysis. In performing these analyses, we considered whether conclusions would have differed if:
-
eligibility had been restricted to studies without high risk of attrition bias; and
-
eligibility had been further restricted to studies that used validated scales to measure vasomotor symptoms.
Overall quality of the body of evidence ‐ Summary of findings table
We used GRADEPRO software and methods of The Cochrane Collaboration to prepare a Summary of findings table (Higgins 2011). This table portrayed the overall quality of the body of evidence for main review outcomes (occurrence of vasomotor symptoms, vaginal bleeding, breast cancer, endometrial cancer, venous thromboembolic events, cardiovascular events, cerebrovascular events and mortality from any cause) and main comparisons (tibolone vs placebo, tibolone vs HT) on the basis of GRADE criteria (study limitations (i.e. risk of bias), consistency of effect, imprecision, indirectness and publication bias). We justified, documented and incorporated Judgements about evidence quality (high, moderate, low or very low) into the reporting of results for each outcome.
Results
Description of studies
See Characteristics of included studies and Characteristics of excluded studies.
Results of the search
The original systematic search performed in 2011 through seven databases produced 540 records (after duplicates were removed). After selecting 57 papers of potential interest from their titles and abstracts, we eventually included 33 RCTs. Two of these articles (Ziaei 2010; Ziaei 2010b) appeared to report different outcomes for the same study; we have amalgamated these and counted them as a single study in the 2016 update.
We performed additional searches in 2015: we initially selected 62 additional abstracts and found 14 additional RCTs, plus another publication (Bots 2006) for one of the studies already included (Langer 2006). (See Figure 1 for study flow.) We have included in this update six studies that were excluded in the previous version of the review (see Differences between protocol and review). Therefore this review update includes a total of 46 studies (32 studies from the previous version of the review, six that were excluded from the previous version of the review and eight new studies).
Of these newly included reports, five (Bouchard 2012; Gupta 2013; Jacobsen 2012; Morais‐Socorro 2012; Polisseni 2013) were published since 2012, and three (Baracat 2002;Doren 1999;Wender 2004) were cited among references provided in other studies. We asked drug manufacturers, as well as authors of conference proceedings, about possibly unpublished studies but obtained no information on this.
Included studies
Study design and setting
We included 46 RCTs of parallel design; 18 were multi‐centre studies.
Participants
All selected RCTs included postmenopausal or perimenopausal women (n = 19,976), and in most of these RCTs, all or some participants had menopausal symptoms. A few studies did not clearly specify whether women were symptomatic, or whether investigators had other reasons to test the effectiveness of tibolone. Among these, five RCTs (Archer 2007; Hänggi 1997; Doren 1999; Okon 2005; Wender 2004) were carried out with the main objective to assess endometrial safety associated with the use of tibolone, and four RCTs (Elfituri 2005; Huber 2002; Winkler 2000; Ziaei 2010) had as their main objective assessment of bleeding patterns.
Five of the included RCTs (Cummings 2008; Gallagher 2001; Jacobsen 2012; Langer 2006; Roux 2002) assessed effects of tibolone on bone loss in postmenopausal women, in addition to its safety profile and its effects on menopausal symptoms. One study (Cummings 2008) also evaluated the reduction in fractures among women with osteoporosis.
Three RCTs (Kenemans 2009; Kroiss 2005; Kubista 2007) specifically studied individuals with breast cancer: Kenemans 2009 assessed the recurrence of breast cancer in women with vasomotor symptoms who were previously treated surgically; Kroiss 2005 evaluated the safety profile of tibolone administered to postmenopausal women after breast cancer surgery to prevent, relieve or delay the occurrence of menopausal symptoms; Kubista 2007 assessed the safety of 14‐day tibolone treatment of breast tissue in patients with invasive cancer without metastatic spread, and we included this study because an ischaemic stroke occurred.
Among populations with specific characteristics other than menopausal symptoms, one RCT (de Aloysio 1998) selected patients with uterine leiomyomas to assess the effects of tibolone on bleeding patterns. Another RCT (Vieira 2009) assessed the frequency of flares in patients with lupus erythematosus.
Most of the included RCTs studied women in natural menopause only, although a few studies also included women without a uterus. In these cases, investigators evaluated endometrial outcomes (bleeding, hyperplasia, cancer) only in women with an intact uterus.
The mean age of women in most of the selected studies was between 52 and 55 years. In two trials (Cummings 2008; Jacobsen 2012) that selected women older than 60 years of age, researchers observed much higher means, whereas in one trial (Elfituri 2005) on Lybian women with natural or surgical menopause, the mean age of participants was lower (44 years). Mean time since menopause ranged from 1.5 to 17 years.
All but three of the selected RCTs included fewer than 1000 participants. Each of the three largest RCTs (Archer 2007; Cummings 2008; Kenemans 2009) actually included more than 3000 participants. Follow‐up periods ranged from two weeks to four years.
Interventions
The included studies administered oral tibolone (usually 2.5 mg daily: range 0.625 mg to 5 mg daily) compared with placebo, unopposed oestrogen or combined HT, as detailed below. Unless otherwise stated, doses were daily and progesterone was continuous. Several studies included more than one comparator.
-
Placebo (17 RCTs): Benedek‐Jaszmann 1987, Berning 2000, Bouchard 2012, Cummings 2008, Gallagher 2001, Hudita 2003, Jacobsen 2012, Kenemans 2009, Kroiss 2005, Kubista 2007, Landgren 2002, Meeuwsen 2002, Morais‐Socorro 2012, Swanson 2006, Vieira 2009, Volpe 1986, Wender 2004
-
Unopposed oestrogen (three RCTs)
-
Conjugated equine oestrogen (CEE) 0.0625 (Gupta 2013)
-
Oestriol 2 to 4 mg (Volpe 1986)
-
17β‐Oestradiol patch 50 μg (Mendoza 2000)
-
-
Combined HT (28 RCTs)
-
CEE 0.625 mg plus medroxyprogesterone acetate 2.5 to 5 mg (Archer 2007;Baracat 2002;de Aloysio 1998;Huber 2002;Kökçü 2000;Langer 2006;Uygur 2005;Wu 2001;Ziaei 2010)
-
Oestradiol valerate 2 mg and norethisterone 0.7 to 2mg (Al‐Azzawi 1999;Okon 2005)
-
Oestradiol 50 μg + norethisterone acetate (140 microgr) in the form of a transdermal patch (Nijland 2009)
-
Oestradiol valerate 2 mg plus dienogest 2 mg (Osmanağaoğlu 2006)
-
Oestradiol 2 mg + oestriol 1 mg/d + norethindrone acetate 1 mg/d (Winkler 2000)
-
Oestradiol 1 to 2 mg plus norethindrone 0.5 to 1 mg (Polisseni 2013;Roux 2002)
-
17β‐Oestradiol 1 to 2 mg + norethisterone 0.5 to 1 mg (Doren 1999;Hammar 1998;Hammar 2007;Nappi 2006a;Nathorst‐Böös 1997)
-
Oestradiol 2 mg + medrogestone 10 mg (Egarter 1996)
-
CEE 0.625 mg plus sequential 150 μg norgestrel (Ross 1999)
-
CEE 0.625 mg plus sequential medroxyprogesterone 5 mg (Siseles 1995)
-
CEE 0.625 mg plus sequential norethisterone 5 mg (Siseles 1995;Volpe 1986)
-
CEE 0.625 mg + sequential cyproterone acetate 12.5 mg/d (Volpe 1986)
-
Oestradiol valerate 2 mg plus sequential cyproterone acetate 12.5 mg (Volpe 1986)
-
Oestradiol valerate 2 mg plus sequential norethisterone 5 mg (Volpe 1986)
-
17β‐Oestradiol oral 2 mg or patch 50 μg plus sequential oral dydrogesterone 10 mg (Elfituri 2005;Hänggi 1997)
-
17β‐Oestradiol patch 50 μg plus sequential norethisterone 0.25 mg (Mendoza 2002)
-
Transdermal β‐oestradiol patch 50 μg plus micronised natural progesterone 200 mg twice a week (Mendoza 2002)
-
Outcomes
Of 46 RCTs, 23 evaluated the effectiveness of tibolone for treatment of vasomotor symptoms in symptomatic women, measured as occurrence (Kökçü 2000;Meeuwsen 2002), as frequency (Bouchard 2012;Hammar 2007;Landgren 2002;Swanson 2006) or with the use of scales (Benedek‐Jaszmann 1987; Elfituri 2005;Hammar 1998;Huber 2002;Hudita 2003;Morais‐Socorro 2012;Polisseni 2013;Wu 2001;Ziaei 2010). Data from eight other RCTs (Al‐Azzawi 1999;Baracat 2002;Egarter 1996;Ross 1999;Siseles 1995;Vieira 2009;Volpe 1986; Wender 2004) that evaluated vasomotor symptoms were unsuitable for analysis (see Table 1 for detailed explanations).
-
Twenty‐eight of 46 RCTs evaluated unscheduled bleeding (24 could be considered for meta‐analyses).
-
Ten of 46 RCTs evaluated breast cancer.
-
Thirteen of 46 RCTs evaluated endometrial cancer.
-
Nine of 46 RCTs evaluated venous thromboembolic events.
-
Five of 46 RCTs evaluated cardiovascular events.
-
Eight of 46 RCTs evaluated cerebrovascular events.
-
Six of 46 RCTs evaluated mortality from any cause.
-
Nine of 46 RCTs evaluated endometrial hyperplasia (extra one is Volpe 1986).
-
Sixteen of 46 RCTs evaluated vaginal dryness and painful sexual intercourse (seven could be considered for meta‐analyses) (extra ones are Mendoza 2000 and Uygur 2005).
-
Four of 46 RCTs evaluated insomnia.
-
Two of 46 RCTs evaluated vaginal infection.
-
One of 46 RCTs evaluated urinary tract infection.
Excluded studies
We excluded 24 studies from the review. Following are the most common reasons for exclusion (occurring in more than one RCT).
-
Three of 24 were not randomised.
-
Fifteen of 24 did not assess outcomes of interest.
-
Four of 24 did not include a comparator of interest.
Risk of bias in included studies
See also Characteristics of included studies,Figure 2 and Figure 3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Sequence generation
Twenty RCTs described adequate methods of sequence generation; we rated them as having low risk of bias in this domain. We rated 25 studies as having unclear risk. We rated one study (Wu 2001) as having high risk of bias; investigators stated they allocated to treatment groups randomly selected pairs of two women.
Allocation concealment
Most of the selected RCTs provided no information regarding allocation concealment. Only 10 of 46 RCTs specified that researchers used a system for concealing allocation (low risk of bias): an interactive voice response system in five RCTs, another computerised system (the Almedica Drug Labelling System; Almedica, Parsippany, NJ, USA) in one RCT and opaque envelopes in four RCTs. We rated remaining studies as having unclear risk of bias.
Blinding
Performance bias
In 22 out of 46 RCTs, participants and/or personnel were blinded (low risk of bias). Fourteen RCTs were open trials or blinding appeared unlikely (high risk of bias), and 10 provided insufficient or no information by which this domain could be assessed (unclear risk).
Detection bias
We considered risk of bias as low in 25 of 46 RCTs, whereas 10 RCTs did not provide enough information for assessment, and we rated 13 studies as having high risk of bias in this domain.
Incomplete outcome data
We considered 17 of 46 RCTs to have low risk of attrition bias. Several RCTs reported some reasons for concern (lack of intention‐to‐treat analysis, loss to follow‐up with no reasons specified). In particular, investigators gave no clear reasons for excluding participants from treatment and/or evaluation in six RCTs (rated as having unclear risk), and more than 10% of participants were lost to follow‐up in 23 RCTs (rated as having high risk).
Selective reporting
Only nine of 46 study protocols were available; we judged risk of selective reporting bias as low in all of these studies, as they reported expected outcomes of interest for this review, or they reported data on adverse events that were not indicated in the study protocol but could be expected in the study report. We rated all other studies as having unclear risk.
Other potential sources of bias
The drug producer sponsored most of the RCTs, and its employees often authored the articles. We rated 26 as having high risk of bias and 10 unclear risk. Just six of 46 RCTs appeared truly independent, and we rated them as having low risk of bias in this domain.
Effects of interventions
See: Summary of findings for the main comparison Tibolone compared with placebo for treatment of vasomotor symptoms in postmenopausal women; Summary of findings 2 Tibolone compared with placebo for postmenopausal women: adverse events; Summary of findings 3 Tibolone compared with combined HT for treatment of vasomotor symptoms in postmenopausal women; Summary of findings 4 Tibolone compared with combined HT for postmenopausal women: adverse events
Tibolone versus placebo
Primary outcomes
Vasomotor symptoms
Eight RCTs reported useable data on this outcome; three other RCTs reported data that could not be used (see Table 1). A substantial effect of tibolone on vasomotor symptoms compared with placebo is suggested (see Analysis 1.1 and Figure 4), with a pooled estimate of the SMD of ‐0.99 (95% CI ‐1.10 to ‐0.89; n = 1657; I2 = 96%; moderate‐quality evidence). Multiplying this by the pooled standard deviation from Hammar 1998 (0.76) suggests that tibolone could improve vasomotor symptoms by around 0.75 (0.7 to 0.8) points on a 5‐point severity scale. A sensitivity analysis (see Analysis 1.15) excluding three RCTs with attrition bias (Benedek‐Jaszmann 1987;Hudita 2003; Morais‐Socorro 2012 ‐ the latter two also have very large estimates) still shows an effect of tibolone, with reduced heterogeneity and effect size (SMD ‐0.61, 95% CI ‐0.73 to ‐0.49; I2 = 54%). The corresponding odds ratio (OR) is 0.33 (95% CI 0.27 to 0.41). These estimates can be translated to meaningful scales; multiplying the SMD by the pooled standard deviation from Hammar 1998 (0.76) suggests that tibolone could improve vasomotor symptoms by around 0.5 (0.4 to 0.6) points on a 5‐point severity scale; this probably would not constitute a clinically meaningful effect.

Forest plot of comparison: 1 Tibolone versus placebo, outcome: 1.1 Vasomotor symptoms.
Subgroup analysis by dose
We found strong evidence (P < 0.00001) of differences between subgroups defined by tibolone dose, although this was diminished when we removed trials with high risk of attrition bias, which were likely to provide overestimates (P = 0.04). Furthermore, once we removed these trials, we noted the suggestion of a dose‐response relationship (Analysis 1.15; Figure 5), although trials were too few to allow formal investigation of this through meta‐regression.

Forest plot of comparison: 1 Tibolone versus placebo, outcome: 1.15 Sensitivity analysis ‐ Vasomotor symptoms without trials with high risk of attrition bias.
Subgroup analysis by duration
We noted some scope, albeit limited, for review authors to consider the impact of treatment duration on the effect; estimates from four of the included studies (Bouchard 2012;Landgren 2002;Morais‐Socorro 2012; Swanson 2006) corresponded to 12 weeks, from one (Hudita 2003) to 24 weeks, from one (Ziaei 2010) to six months and from one (Benedek‐Jaszmann 1987) to 12 months. All seven studies appeared in the stratum corresponding to a dose of 2.5 mg/d. Accordingly, we were able to look at estimates in this stratum to see whether duration modified the treatment effect when dose was held constant. As we recalled the high risk of attrition bias in Hudita 2003 and Morais‐Socorro 2012, we noted that no such relationship was evident; neither the estimate from Benedek‐Jaszmann 1987 (12 months) nor that from Ziaei 2010 (six months) was notably different from the 12 week estimates.
Unscheduled bleeding
Nine RCTs reported this outcome (Analysis 1.2). Unscheduled bleeding was more likely to occur in the tibolone group (OR 2.79, 95% CI 2.10 to 3.70; nine RCTs; n = 7814; I2 = 43%; moderate‐quality evidence). This suggests that if 18% of women taking placebo experience unscheduled bleeding, then between 31% and 44% of women taking tibolone will do so. Statistical significance persisted if we excluded the two largest RCTs (Cummings 2008; Kenemans 2009), which provided 47% of the total weight and about 85% of the population of interest.
Subgroup analysis by dose
Results were stratified by dose (2.5 and 1.25 mg daily). Effect estimates were similar in the two groups.
Long‐term adverse events
Endometrial cancer
Eight RCTs reported this outcome (Analysis 1.3). We found no evidence of a difference between groups, although the event rate was low, with 16 cases reported in the tibolone arms and five in the placebo arms (OR 2.04, 95% CI 0.79 to 5.24; eight RCTs; 8504 women; I2 = 0%; very low‐quality evidence).
Evidence suggests that if one woman in a thousand taking placebo develops endometrial cancer, then between one and six women in a thousand who take tibolone may do so. Seven and four cases, respectively, occurred in Kenemans 2009 (with 2.5 mg/d; n = 3133), and four versus zero cases in Cummings 2008 (with 1.25 mg; n = 3519). Fifteen cases (11 in tibolone arms vs four in placebo arms) occurred in studies recruiting younger postmenopausal women (average age < 55 years).
Breast cancer
Six RCTs assessed this outcome: four in women without a history of breast cancer (Analysis 1.4) and two in women with a history of breast cancer (Analysis 1.5).
Among women without a history of breast cancer, we found no evidence of a difference between groups (OR 0.52, 95% CI 0.21 to 1.25; four RCTs; 5500 women; I2 = 17%; very low‐quality evidence).
Among women with a history of breast cancer, we noted increased risk in the tibolone group (OR 1.5, 95% CI 1.21 to 1.85; two RCTs; 3165 women; moderate‐quality evidence). All events occurred in the largest of the studies (Kenemans 2009), which administered 2.5 mg/d of tibolone and was stopped prematurely owing to increased risk in the intervention group.
Venous thromboembolic events
Five RCTs assessed this outcome; three of them (Cummings 2008; Kenemans 2009;Landgren 2002) reported the occurrence of events (Analysis 1.6). We found no evidence of a difference between groups (OR 0.85, 95% CI 0.37 to 1.97; n = 9176; I2 = 0%; very low‐quality evidence).
Ten cases (seven in tibolone arms vs three in placebo arms) of a total of 24 occurred in studies recruiting younger postmenopausal women (average age < 55).
Cardiovascular events
We found no evidence of a difference between groups (OR 1.38, 95% CI 0.84 to 2.27; four RCTs; n = 8401; I2 = 0%; very low‐quality evidence; Analysis 1.7).
The four RCTs assessing this outcome involved women of very different age groups (Cummings 2008, mean age 68; Jacobsen 2012, mean age 74; Kenemans 2009, mean age 53 years; Langer 2006, mean age 59), but we observed no statistical heterogeneity between these studies.
Cerebrovascular events
Four RCTs assessed this outcome (Analysis 1.8) and provided no conclusive evidence of a difference between groups (OR 1.74, 95% CI 0.99 to 3.04; four RCTs; n = 7930; I2 = 0%).
One RCT (Cummings 2008; n = 4506), which selected osteoporotic women aged 60 to 85 years, provided most of the data; this trial was stopped prematurely for increased risk of stroke with 1.25 mg/d of tibolone (28 vs 13 cases; OR 2.18, 95% CI 1.12 to 4.21). Among women younger than 60 years old (Kenemans 2009), five cases occurred in each group (OR 0.99, 95% CI 0.29 to 3.42; n = 3133).
Mortality from any cause
Four RCTs assessed this outcome, and three reported events (Analysis 1.9), providing no evidence of a difference between groups (OR 1.06, 95% CI 0.79 to 1.41; five RCTs; n = 8242; I2 = 0%; low‐quality evidence).
Secondary outcomes
Insomnia
Three RCTs reported insomnia or "sleep" (Analysis 1.10).
Results suggested an advantage of tibolone over placebo related to insomnia or quality of sleep (SMD ‐0.19, 95% CI ‐0.38 to 0.00; three RCTs; n = 3432; I2 = 0%).
Genital symptoms
Vaginal dryness
Three RCTs (Hudita 2003;Kenemans 2009;Ziaei 2010) reported useable data on this outcome (see Analysis 1.11 and Table 2), suggesting an advantage of tibolone over placebo for vaginal dryness, although this would barely be evident if the two arms from Hudita 2003, which had a high dropout rate, were excluded. The SMD (95% CI) including Hudita 2003 was ‐0.66 (‐0.90 to ‐0.43), which corresponds to improvement on a 0 to 3 severity score of 0.6 (0.4 to 0.8) points with a standard deviation (SD) of 0.89. This probably would not amount to a clinically meaningful difference.
Vaginal infection
Two RCTs reported this outcome (Analysis 1.12). The rate of vaginal infection was higher in the tibolone group (OR 2.50, 95% CI 1.24 to 5.06; two RCTs; n = 7639; I2 = 88%). The direction of effect was consistent, but considerable statistical heterogeneity was probably due to differences in the population studied (osteoporotic women aged 60 to 85 years in Cummings 2008, and younger women who had experienced breast cancer in Kenemans 2009).
Urinary tract infection
One RCT (Kenemans 2009) reported this outcome (Analysis 1.13) and revealed no evidence of a difference between groups (OR 0.70, 95% CI 0.46 to 1.06; one RCT; n = 3133).
Endometrial hyperplasia
Four RCTs assessed this outcome, and two reported events (Analysis 1.14), providing no evidence of a difference between groups, although results revealed only seven events in total (OR 1.20, 95% CI 0.23 to 6.25; n = 4518; I2 = 0%).
Tibolone versus oestrogens
Primary outcomes
Two RCTs (Gupta 2013;Mendoza 2002) compared tibolone versus oestrogens and reported data on three outcomes (vasomotor symptoms, vaginal dryness and painful sexual intercourse, insomnia).
Vasomotor symptoms
We found no evidence of a difference between groups (OR 1.23, 95% CI 0.35 to 4.34; two RCTs; n = 108; I2 = 0%; low‐quality evidence), although the small number of events observed meant that large effects in either direction could not be ruled out. See Analysis 2.1 and Figure 6.
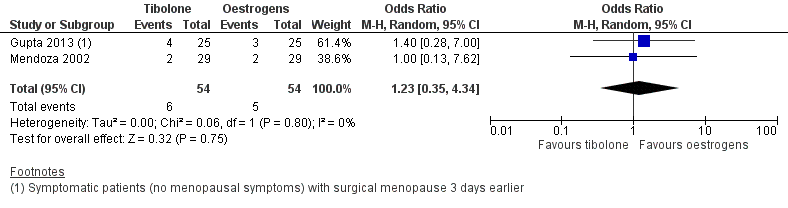
Forest plot of comparison: 2 Tibolone versus oestrogens, outcome: 2.1 Vasomotor symptoms.
Secondary outcomes
Insomnia
No events occurred in either group (Analysis 2.2).
Genital symptoms
Vaginal dryness and painful sexual intercourse
We found no evidence of a difference between groups (OR 0.32, 95% CI 0.01 to 8.25; one RCT; n = 50), although the estimate was so imprecise as to be completely uninformative (Analysis 2.3).
Tibolone versus combined HT
Primary outcomes
Vasomotor symptoms
Nine RCTs reported useable data on this outcome, and five other RCTs provided data that could not be used (see Table 1). Results suggested a small disadvantage of tibolone compared with combined HT (see Analysis 3.1 and Figure 7), with a pooled estimate of the SMD of 0.17 (95% CI 0.06 to 0.28; n = 1336; I2 = 67%; moderate‐quality evidence). Multiplying this estimate by the pooled standard deviation from Hammar 1998 (0.76) suggests that combined HT improves vasomotor symptoms by around 0.15 (0.08 to 0.23) compared with tibolone on a 5‐point severity scale. The corresponding OR was 1.36 (95% CI 1.11 to 1.66). A sensitivity analysis (see Analysis 3.11) excluding five RCTs with high attrition bias provided slightly larger but similar estimates (SMD 0.25, 95% CI 0.09 to 0.41; I2 = 0%). A further sensitivity analysis excluding the latter five RCTs plus Hammar 1998 (using a non‐validated scale) revealed no evidence of a difference between treatments because the estimate lacked precision once other studies were excluded (see Analysis 3.12).

Forest plot of comparison: 3 Tibolone versus combined HT, outcome: 3.1 Vasomotor symptoms.
Subgroup analysis by duration
Duration of treatment in this comparison ranged from 12 weeks to 12 months, while dose was the same in all studies (2.5 mg/d); therefore, a tentative investigation of the impact of treatment duration on treatment effect could be undertaken. Although we identified too few studies to permit a formal analysis (e.g. using meta‐regression), we were able to order the studies according to duration so as to inspect whether a trend in the size of the SMDs was suggested (Analysis 3.13). However, we observed no clear trend, and consequently found no evidence that the difference between tibolone and HT varies according to the duration of treatment.
Unscheduled bleeding
Seventeen RCTs reported this outcome: 15 compared tibolone with continuous combined HT, two with continuous sequential HT (Analysis 3.2). The latter studies included cases of bleeding if they had been reported as side effects by study authors.
Tibolone was associated with fewer breakthrough events than combined HT (OR 0.32, 95% CI 0.24 to 0.41; 16 RCTs; n = 6438; I2 = 72%; low‐quality evidence), suggesting that if 47% of women taking combined HT experience unscheduled bleeding, then between 18% and 27% of those taking tibolone will do so. High heterogeneity was attributable in part to an RCT (Nijland 2009) in which HT was delivered in patch form, and also to a difference between dose subgroups, as noted below.
Statistical significance persisted if we excluded the largest RCT (Archer 2007, which provided about half of the population of interest).
One RCT (Okon 2005) reported this outcome as days of bleeding over one year of follow‐up. Study authors reported no significant differences between groups.
Subgroup analysis by dose
We stratified results by dose, revealing a statistically significant difference between 2.5 mg and 1.25 mg subgroups (test for subgroup differences: Chi² = 7.28; df = 1 (P = 0.007); I² = 86.3%), which suggested that the lower dose of tibolone was associated with a more beneficial effect when compared with HT (OR 0.21, 95% CI 0.16 to 0.26; two RCTs; n = 1718; I2 = 0%).
Long‐term adverse events
Endometrial cancer
Five RCTs reported this outcome (Analysis 3.3). Few events occurred (two cases in tibolone arms vs one in combined HT arms in three trials), and investigators provided no evidence of a difference between groups (OR 1.47, 95% CI 0.23 to 9.33; five RCTs; n = 3689; I2 = 0%; very low‐quality evidence).
Breast cancer
Five RCTs assessed this outcome (Analysis 3.4). All included women without a history of breast cancer. Few events occurred (17 cases in tibolone arms vs 10 in combined HT arms), and researchers provided no evidence of a difference between groups (OR 1.69, 95% CI 0.78 to 3.67; n = 4835; I2 = 0%; very low‐quality evidence).
Twenty‐two cases (13 in tibolone arms vs nine in placebo arms) occurred in studies recruiting younger postmenopausal women (average age < 55).
Venous thromboembolic events
Four RCTs assessed this outcome (Analysis 3.5). Few events occurred (one case of pulmonary embolism in tibolone arms vs two cases of pulmonary embolism and three of deep venous thrombosis in combined HT arms), and researchers provided no evidence of a difference between groups (OR 0.44, 95% CI 0.09 to 2.14; four RCTs; n = 4529; I2 = 0%; very low‐quality evidence).
Cardiovascular events
Two RCTs assessed this outcome (Archer 2007; Langer 2006). Few events occurred (seven in tibolone arms vs 11 in combined HT arms), and results showed no evidence of a difference between groups (OR 0.63, 95% CI 0.24 to 1.66; two RCTs; n = 3794; I2 = 0%; very low‐quality evidence; Analysis 3.6). The mean age of women in these RCTs was less than 60 years.
Cerebrovascular events
Four RCTs assessed this outcome (Analysis 3.7). Few events occurred (two cases in tibolone arms vs four cases in combined HT arms), and data show no evidence of a difference between groups (pooled OR 0.76, 95% CI 0.16 to 3.66; four RCTs; n = 4562; I2 = 0%; very low‐quality evidence). The mean age of women in these RCTs was less than 60 years.
Mortality from any cause
Two RCTs (Langer 2006; Nijland 2009; n = 970) reported this outcome, with only one case noted in the tibolone arm (Analysis 3.8).
Secondary outcomes
Insomnia
Just one RCT (Egarter 1996) used a validated scale (a domain of the Kupperman Index) to assess this outcome but provided no data suitable for analysis (SD was not reported and could not be calculated sensibly via the information provided). The publication reported no evidence of a difference between tibolone and combined HT.
Genital symptoms
Vaginal dryness and painful sexual intercourse
Evidence at face value suggested little or no difference between tibolone and combined HT in relation to vaginal dryness (SMD 0.02, 95% CI ‐0.12 to 0.17; seven RCTs; n = 1098; moderate‐quality evidence; Analysis 3.10).
Mendoza 2000 (n = 76) also measured painful sexual intercourse as an outcome but provided no data suitable for analysis; study authors reported no significant difference between groups.
Similarly, Nathorst‐Böös 1997 evaluated dyspareunia but provided no data suitable for analysis, and study authors reported that they found no evidence of a difference between groups.
Vaginal infection
None of the selected RCTs reported useable data on this outcome
Urinary tract infection
None of the selected RCTs reported useable data on this outcome.
Endometrial hyperplasia
Five RCTs assessed this outcome (Analysis 3.9), reporting few events (zero cases in tibolone arms vs three cases in the combined HT arm) and no evidence of a difference between groups (OR 0.35, 95% CI 0.05 to 2.21; five RCTs; n = 2846; I2 = 0%).
Sensitivity analyses
Aside from sensitivity analyses performed for evaluation of vasomotor symptoms, as described above (see Results 1.1 and 3.1), review authors performed sensitivity analyses for primary outcomes, considering alternative scenarios in participants lost to follow‐up. We performed three analyses on placebo‐controlled RCTs (specifically on venous thromboembolic events and breast cancer in women who had or had no history of breast cancer) and two on combined HT controlled RCTs (specifically on unscheduled bleeding and vasomotor symptoms). None of these analyses showed differences in terms of direction of effect and statistical significance.
Assessment of review‐wide reporting bias
Funnel plot analyses were not helpful to review authors in assessing the presence of publication bias, given the relative scarcity of studies and data. Vasomotor symptoms and unscheduled bleeding were the only outcomes with sufficient RCTs to permit such an assessment, which revealed no evidence of bias for this outcome. As for the other outcomes, we cannot exclude the occurrence of publication bias because the drug manufacturer, who sponsored almost all of the published RCTs, was asked for possibly unpublished data but provided no written response.
Discussion
Summary of main results
For this review, we retrieved randomised controlled trials (RCTs) comparing tibolone versus placebo and versus combined hormone therapy (HT). We identified only three RCTs comparing tibolone versus oestrogens without progestogens (Gupta 2013; Mendoza 2000; Volpe 1986), and only two of these were suitable for analysis. The addition of progestogens is considered important for lowering the risk of endometrial carcinoma in women with a uterus.
Effectiveness in treatment of menopausal symptoms
Our findings suggest that tibolone reduces vasomotor symptoms compared with placebo and is less effective than combined HT. The clinical relevance of observed differences is disputable ‐ especially for comparison versus combined HT ‐ as their magnitude is limited. It should be noted that the quality of evidence for this outcome was moderate. In particular, attrition bias and use of non‐validated scales were frequently observed, as was statistical heterogeneity, although sensitivity analyses excluding RCTs with high risk of attrition bias confirmed both statistical significance and direction of effects. Available evidence suggests at most a modest effect of tibolone on insomnia and vaginal dryness compared with placebo. No clinically relevant differences are apparent between tibolone and combined HT in relation to vaginal dryness outcomes.
Short‐term safety
This review suggests that tibolone has a better bleeding profile than combined HT and is associated with more numerous breakthrough bleeding events than placebo.
Evidence is scarce and unclear on vaginal and urinary tract infections. Only two RCTs (Cummings 2008; Kenemans 2009) provided data on vaginal infection. Cummings 2008 performed cervical cytological smears annually in women with a cervix, whereas Kenemans 2009 provided no information on diagnostic technique. Both RCTs suggested that tibolone increases vaginal infection and provided no information on specific aetiologic agents. Only one study reported urinary tract infections.
Long‐term safety
For this systematic review, we found few RCTs providing data that could be used to assess the long‐term safety of tibolone. Nearly all of the evidence on adverse events was of very low quality, and events were scarce.
Available evidence indicates that compared with placebo, tibolone increases the risk of recurrent breast cancer in women with a history of breast cancer, and may increase the risk of stroke among women over 60 years of age. No evidence suggests that tibolone increases the risk of other long‐term adverse events, and no evidence reveals a difference between tibolone and HT with respect to long‐term adverse events.
In particular, the LIBERATE study (Kenemans 2009) confirmed that tibolone could significantly increase breast cancer among high‐risk women who were surgically treated within five years for breast cancer (for whom usual oestrogen and combined HT therapies were contraindicated) and who were using adjuvant therapy and/or chemotherapy in about seven cases out of 10. A daily dose of 2.5 mg led to an average of 15 extra recurrences each year for every 1000 women. It is a matter of concern that more than 70% of recurrence events were distant metastases, ultimately leading to death. This study failed to confirm the initial hypothesis of non‐inferiority of tibolone versus placebo for breast cancer risk, and was stopped after 3.1 years.
The latter findings sharply contrast with results from the LIFT study (Cummings 2008), in which 1.25 mg of tibolone, administered to osteoporotic women to reduce the risk of vertebral fracture, slightly but significantly reduced new‐onset breast cancer (about two fewer cases for every 1000 women each year). However, the absolute number of events in this study was low (six for tibolone vs 19 for placebo, for a total population of about 4500 women between 60 and 85 years of age). We should also note that LIFT researchers used half of the recommended dose for menopausal symptoms in women over 60 years of age (mean age 68). The Million Women Study (Beral 2011) suggested that breast cancer risk may be greater in women starting hormonal therapies within five years of menopause.
Populations for the LIBERATE and LIFT studies were too different for results to be combined meaningfully, and populations in both studies are not a typical target for HT addressing menopausal symptoms, so transferability of their results is a matter of concern. Other RCTs have not added useful data for better assessment of the breast cancer hypothesis. We should consider that follow‐up in available RCTs was between 12 weeks and three years, which may be too short a period for a drug therapy to induce cancer, except for the LIBERATE study, in which high‐risk women were treated and the study was powered for assessment of breast cancer recurrence.
We found 13 RCTs reporting on endometrial cancer, which occurred in only seven of these trials. Its incidence was low (most cases occurred in placebo‐controlled trials ‐ 15 cases in tibolone arms vs five cases in placebo arms ‐ most in Kenemans 2009), so that the hypothesis emerging from observational studies of greater risk with tibolone could not be confirmed. In this case, we should also consider that study follow‐up ranged between 12 weeks and three years ‐ an inadequate duration for a drug therapy to induce cancer.
Data on cerebrovascular events provide some suggestion of higher risk of stroke with tibolone versus placebo. This result was driven by the LIFT study (Cummings 2008), which recruited women over 60 years of age and stopped after 33 months for such an unexpected difference of 2.3 more events every 1000 women per year, which was even greater during the first year of treatment. These data are consistent with data from systematic reviews of RCTs testing combined HT therapies versus placebo; among those, a Cochrane review (Sanchez 2005) including 10 RCTs with a total of 24,283 women randomised to hormone therapy (HT) or placebo for an average of five years (risk ratio (RR) for stroke 1.25, 95% confidence interval (CI) 1.07 to 1.45). As for RCTs directly comparing tibolone versus combined HT, our review did not show differences between treatments, but data were scant. Unpublished data from the Million Women Study (available as rapid response; Beral 2007) had suggested higher risk of fatal stroke with tibolone versus other hormonal therapies (RR 1.58, 95% CI 1.06 to 2.37).
Our review provides no evidence of an increase in cardiovascular events with tibolone versus placebo, whereas data on thromboembolic events are very scant and unhelpful. As for combined HT, Sanchez 2005 found no increase in cardiovascular events and total mortality with HT but reported an increase in thromboembolic events. Randomised controlled trials directly comparing tibolone versus combined HT have provided few data and have revealed no statistically significant differences.
Last, two large RCTs (Cummings 2008; Kenemans 2009), which included higher‐risk women than were included in other studies (for previous cancer or more advanced age), provided most of the data on mortality, revealing no statistically significant differences or trends.
Summary of benefits and harms
Moderate‐quality evidence suggests that tibolone is more effective than placebo and less effective overall than combined HT in reducing postmenopausal symptoms, although the magnitude of observed differences is low. Tibolone provides a clear advantage in terms of less vaginal bleeding, but available data from RCTs on its long‐term safety compared with other hormonal therapies are insufficient.
We found no evidence that tibolone increases the risk of serious adverse events for women taking it over a short term to treat vasomotor symptoms, provided they have had no history of breast cancer, but data are scarce and more evidence is required. Evidence indicates that tibolone is associated with increased risk of serious adverse events when used in other contexts. Tibolone leads to increased risk of breast cancer among women with a history of breast cancer and appears to increase the risk of stroke in older women. Data on endometrial cancer are inconclusive.
Overall completeness and applicability of evidence
Moderate‐quality evidence on symptomatic relief may limit its applicability and clinical relevance. Very little evidence is available on the risks of breast and endometrial cancer in women typically treated for menopausal symptoms. In addition to this, we found no unpublished studies and did not obtain such information from the drug manufacturer. It should be highlighted that absence of publication bias is unusual in therapeutic areas with strong commercial interests, especially as almost all of the published RCTs were sponsored by the drug manufacturer (Bekelman 2003; Lexchin 2003).
Most of the included RCTs assessed effects of tibolone 2.5 mg ‐ the most frequently used dose. Therapeutic schemes and doses of active controls (combined HT) also reflect those normally used. Most of the selected RCTs included postmenopausal women with menopausal symptoms. Two of the largest RCTs, which strongly influenced results on several outcomes, included very specific populations (patients with breast cancer and those with osteoporosis, respectively), and findings of these studies are of limited applicability to women taking tibolone for menopausal symptoms.
Quality of the evidence
We rated the quality of the evidence for the primary outcome of our review ‘vasomotor symptoms’ as moderate for comparisons of tibolone versus placebo and combined HT, and very low for the comparison against oestrogens. We consider the quality to be very low for the comparison versus oestrogen because we identified only two small studies, both of which were compromised by attrition bias. Given that dropout in these studies is very likely to be informative (women with poorer responses will be more likely to drop out), attrition could be fatal to the validity of a trial. In relation to comparisons against combined HT and placebo, we have identified weaknesses in many of the individual studies. However, on the basis of our sensitivity analyses, we believe we can be reasonably confident in our conclusions related to vasomotor symptoms, for the following reasons.
First, many of the relevant studies in these comparisons are subject to attrition bias, which, as noted above, could undermine the validity of a trial. However, we have shown that our conclusions are quite robust if we include only studies without high risk of attrition bias. Another concern is the matter of poor reporting in these studies. This is a matter of concern because we had to make some assumptions about variance in some studies, and we had to pool outcomes measured on different scales. However, although this may have had some impact on the exact size (and precision) of the estimate, it is probably unlikely that we arrived at estimates in the wrong direction (i.e. it is unlikely that placebo is actually better than tibolone, or that HT is worse than tibolone, with respect to vasomotor symptoms). Heterogeneity among studies is notable, but for the comparison versus placebo, we appear to explain much of it as the result of dose effects and artificially large estimates due to attrition bias in several studies. Substantial heterogeneity remains for the comparison versus HT, which we cannot explain; we see no evidence of a difference in treatment effectiveness according to treatment duration, and considerable variation remains after studies with high risk of attrition bias were excluded. One study (Hammar 1998) dominates this comparison: It is reasonably sized and appears to be of fair quality (given its use of a non‐validated measurement scale). This study has a conflict of interest, as the manufacturer of tibolone is involved. However, the estimate from this trial actually suggests a disadvantage of tibolone, so the conflict of interest is not really a concern. Many of the other included studies have similar conflicts of interest. However, specific concerns in relation to this would involve selective reporting and publication bias, and we would expect these to manifest as artificial exaggeration of the benefits of tibolone. We have ended up concluding that tibolone is inferior to HT in relation to vasomotor symptoms; it seems unlikely that companies would be hiding studies or analyses that showed tibolone as superior to HT, so it is unlikely that our conclusion would change if we discovered new studies. These biases may have affected our estimate of the effect of tibolone compared with placebo, although we tentatively note that trials with no apparent conflict of interest also demonstrated benefit in relation to vasomotor symptoms (tentatively, because these studies are themselves subject to other sources of bias). In summary, although the individual studies have weaknesses, we believe we can be fairly confident in our conclusions related to vasomotor symptoms, given the collective evidence. Although the exact size and precision of our estimates could change in light of further research, we believe that our clinical conclusions are reasonably unlikely to do so. In our view, this warrants a GRADE assessment of moderate quality.
We would similarly assess the quality of the evidence for the outcome unscheduled bleeding. We found no evidence for the comparison against oestrogens, but we would consider the evidence to be of moderate quality when taken collectively for the comparisons against placebo and combined HT, because estimates from studies with conflicts of interest and showing attrition bias appear to be generally similar to those from studies not revealing these weaknesses. We have rated the quality of evidence related to other adverse events as very low, as the result of low or very low event rates, leading to imprecision in our estimates and a corresponding inability to comment on the effects of tibolone on these endpoints.
Potential biases in the review process
As stated above, we asked the drug manufacturer, which sponsored almost all of the published RCTs, to provide possibly unpublished data but received no written response. Funnel plot analyses did not help review authors in assessing the presence of publication bias, given the relative scarcity of studies and data, although we were able to produce such plots for both unscheduled bleeding and vasomotor symptoms, and these suggested no obvious bias.
Agreements and disagreements with other studies or reviews
Use of tibolone for the treatment of menopausal symptoms has never been supported by demonstrated advantages over oestrogens and combined HT therapies, such as lower risks of breast and endometrial cancer. On the contrary, observational data from the Million Women Study (Beral 2003; Beral 2005) suggested greater risk of breast cancer (RR 1.45, 95% CI 1.25 to 1.68) and endometrial cancer (RR 1.79, 95% CI 1.43 to 2.25) versus non‐users of HT, and two more recent RCTs included in this review (Cummings 2008; Kenemans 2009) have raised concerns about the benefit/risk profile of this drug. The latter two trials targeted very specific populations (women over 60 years of age and women who had already had breast cancer), and their results are not easily generalisable, although it may be wise to apply a precautionary principle and not exclude the possibility of safety problems for other groups. It should be noted that the Food and Drug Administration rejected the application for the registration of tibolone in the United States, although the reason for this is unknown.
With regard to the effectiveness of tibolone for treating menopausal symptoms, the effectiveness of combined HT over placebo has been shown more convincingly (MacLennan 2004).
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Forest plot of comparison: 1 Tibolone versus placebo, outcome: 1.1 Vasomotor symptoms.
Forest plot of comparison: 1 Tibolone versus placebo, outcome: 1.15 Sensitivity analysis ‐ Vasomotor symptoms without trials with high risk of attrition bias.
Forest plot of comparison: 2 Tibolone versus oestrogens, outcome: 2.1 Vasomotor symptoms.
Forest plot of comparison: 3 Tibolone versus combined HT, outcome: 3.1 Vasomotor symptoms.

Comparison 1 Tibolone versus placebo, Outcome 1 Vasomotor symptoms.

Comparison 1 Tibolone versus placebo, Outcome 2 Unscheduled bleeding.
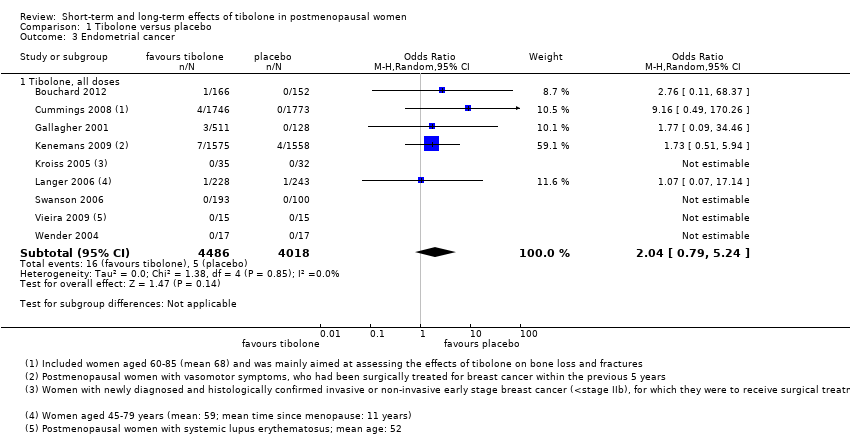
Comparison 1 Tibolone versus placebo, Outcome 3 Endometrial cancer.
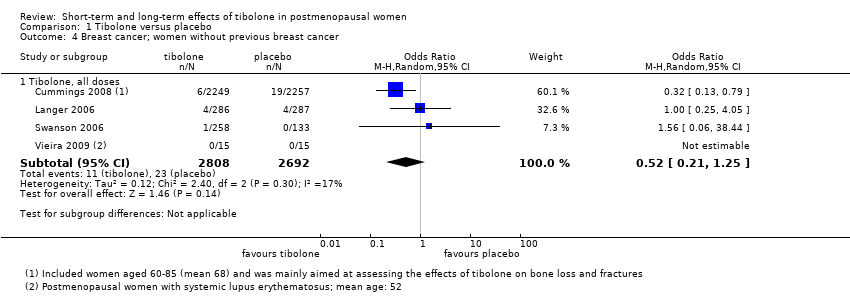
Comparison 1 Tibolone versus placebo, Outcome 4 Breast cancer; women without previous breast cancer.

Comparison 1 Tibolone versus placebo, Outcome 5 Breast cancer; women with previous breast cancer.

Comparison 1 Tibolone versus placebo, Outcome 6 Venous thromboembolic events (clinical evaluation).

Comparison 1 Tibolone versus placebo, Outcome 7 Cardiovascular events.
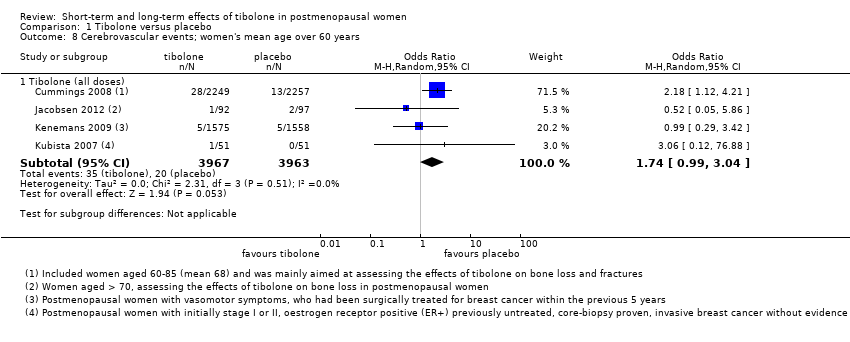
Comparison 1 Tibolone versus placebo, Outcome 8 Cerebrovascular events; women's mean age over 60 years.
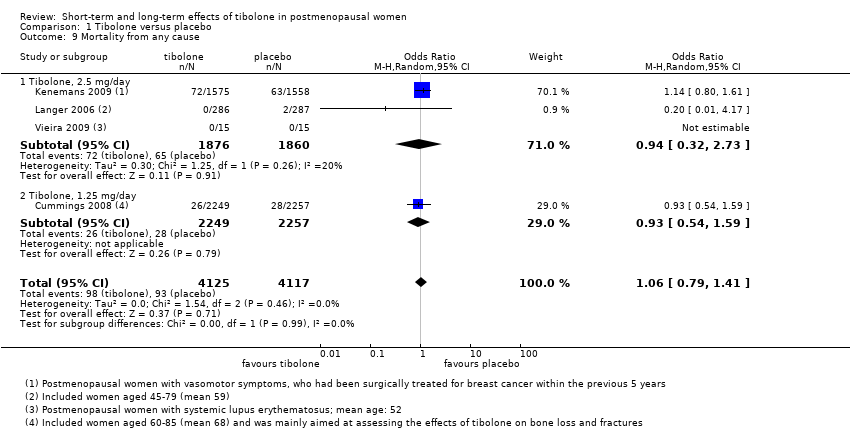
Comparison 1 Tibolone versus placebo, Outcome 9 Mortality from any cause.

Comparison 1 Tibolone versus placebo, Outcome 10 Insomnia.

Comparison 1 Tibolone versus placebo, Outcome 11 Vaginal dryness and painful sexual intercourse.

Comparison 1 Tibolone versus placebo, Outcome 12 Vaginal infections.

Comparison 1 Tibolone versus placebo, Outcome 13 Urinary tract infections.

Comparison 1 Tibolone versus placebo, Outcome 14 Endometrial hyperplasia.

Comparison 1 Tibolone versus placebo, Outcome 15 Sensitivity Analysis ‐ Vasomotor symptoms without trials with high risk of attrition bias.

Comparison 2 Tibolone versus oestrogens, Outcome 1 Vasomotor symptoms.

Comparison 2 Tibolone versus oestrogens, Outcome 2 Insomnia.
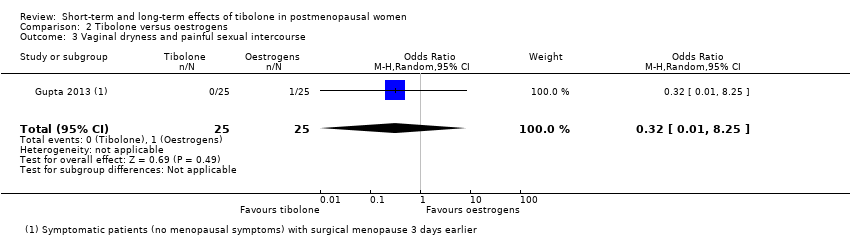
Comparison 2 Tibolone versus oestrogens, Outcome 3 Vaginal dryness and painful sexual intercourse.

Comparison 3 Tibolone versus combined HT, Outcome 1 Vasomotor symptoms.

Comparison 3 Tibolone versus combined HT, Outcome 2 Unscheduled bleeding.

Comparison 3 Tibolone versus combined HT, Outcome 3 Endometrial cancer.

Comparison 3 Tibolone versus combined HT, Outcome 4 Breast cancer; women without previous breast cancer.

Comparison 3 Tibolone versus combined HT, Outcome 5 Venous thromboembolic events (clinical evaluation).
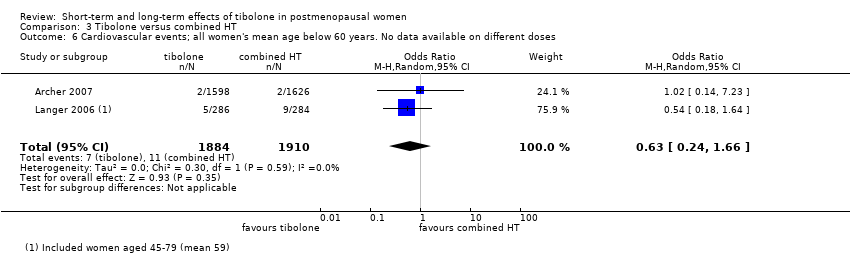
Comparison 3 Tibolone versus combined HT, Outcome 6 Cardiovascular events; all women's mean age below 60 years. No data available on different doses.

Comparison 3 Tibolone versus combined HT, Outcome 7 Cerebrovascular events; women's mean age below 60 years.

Comparison 3 Tibolone versus combined HT, Outcome 8 Mortality from any cause.

Comparison 3 Tibolone versus combined HT, Outcome 9 Endometrial hyperplasia.

Comparison 3 Tibolone versus combined HT, Outcome 10 Vaginal dryness and painful sexual intercourse.
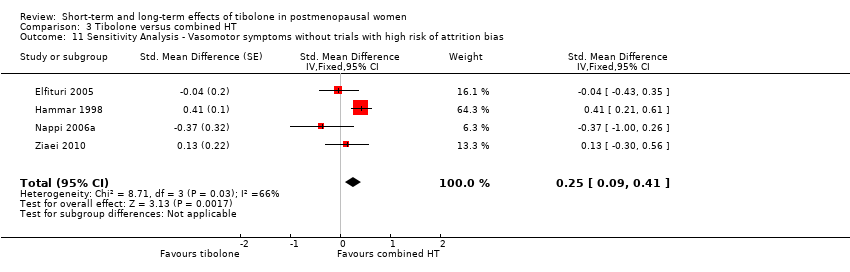
Comparison 3 Tibolone versus combined HT, Outcome 11 Sensitivity Analysis ‐ Vasomotor symptoms without trials with high risk of attrition bias.

Comparison 3 Tibolone versus combined HT, Outcome 12 Sensitivity analysis ‐ vasomotor symptoms ‐ excluding studies with attrition bias and using nonvalidated scales.

Comparison 3 Tibolone versus combined HT, Outcome 13 Vasomotor symptoms ‐ ordered by duration.
| Tibolone compared with placebo: vasomotor symptoms | ||||||
| Population: postmenopausal women with vasomotor symptoms | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Tibolone | |||||
| Vasomotor symptoms (all doses) Follow‐up: 12 weeks to 1 year | 670 per 1000 | 400 per 1000 (350 to 450) | OR 0.33 | 842 (5 RCTs) | ⊕⊕⊝⊝ | Three studies at high risk of attrition bias were excluded from this analysis. Inclusion of these studies was associated with stronger effect of tibolone but with extreme heterogeneity (I2= 97%) |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level for serious risk of bias: poor reporting of study methods and potential conflict of interest (pharmaceutical funding) in most studies; standard deviations imputed for some studies. Effect estimate robust to a sensitivity analysis excluding studies at high risk of attrition bias | ||||||
| Tibolone compared with placebo: adverse events | ||||||
| Population: postmenopausal women with or without vasomotor symptoms | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Tibolone | |||||
| Endometrial cancer (all doses) | See comment | OR 2.04 | 8504 | ⊕⊝⊝⊝ | Events very rare in both groups. Total of 21 events: 16/4486 in tibolone group, 5/4018 in placebo group | |
| Breast cancer; women without previous breast cancer (all doses) | 4 per 1000 | 1 per 1000 (1 to 5) | OR 0.52 | 5500 | ⊕⊝⊝⊝ | In women with a history of breast cancer, risk increased in the tibolone group at 1 to 2.75 years' follow up: OR 1.50 (1.21 to 1.85, 2 RCTs, 3165 women, moderate‐quality evidence ) |
| Unscheduled bleeding (all doses) Follow‐up: 1 to 3 years (median 2) | 177 per 1000 | 374 per 1000 (310 to 442) | OR 2.79 | 7814 | ⊕⊕⊝⊝ | |
| Venous thromboembolic events (clinical evaluation) all doses | See comment | OR 0.85 | 9176 | ⊕⊝⊝⊝ | Events very rare in both groups. Total of 24 events: 12/5054 in tibolone group, 12/4122 in placebo group | |
| Cardiovascular events (all doses) Follow‐up: 2 to 3 years (median 2.75) | 10 per 1000 | 13 per 1000 (8 to 22) | 1.38 (0.84 to 2.27) | 8401 (4 studies) | ⊕⊝⊝⊝ | |
| Cerebrovascular events (all doses) | 5 per 1000 | 8 per 1000 (4 to 14) | OR 1.74 | 7930 | ⊕⊝⊝⊝ | |
| Mortality from any cause (all doses) | 10 per 1000 | 10 per 1000 (8 to 14) | OR 1.06 | 8242 | ⊕⊕⊝⊝ | |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded two levels for very serious risk of bias: poor reporting of study methods, high attrition and/or potential conflict of interest in most studies bDowngraded one level for serious imprecision: low event rate. Findings compatible with meaningful benefit in one or both arms, or with no effect cDowngraded one level for serious risk of low applicability: Some studies compare doses of tibolone that have not been marketed (although downgrading has no effect on rating, as study already rated very low) dDowngraded one level for serious risk of bias: poor reporting of study methods and potential conflict of interest in most studies eDowngraded one level for potential conflict of interest (funding by pharmaceutical companies) | ||||||
| Tibolone compared with combined HT for postmenopausal women: vasomotor symptoms | ||||||
| Population: postmenopausal women with vasomotor symptoms | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Combined HT | Tibolone | |||||
| Vasomotor symptoms (tibolone 2.5 mg/d) | 70 per 1000 | 110 per 1000 (80 to 140) | OR 1.57 | 646 (4 studies) | ⊕⊝⊝⊝ | From a sensitivity analysis excluding studies with high risk of attrition bias. An inclusive analysis (9 studies, 1336 participants) suggests a similar but slightly reduced disadvantage of tibolone (OR (95% CI) 1.36 (1.11 to 1.66)) |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level for serious risk of bias: poor reporting of study methods and potential conflict of interest in all studies. Effect estimate robust to a sensitivity analysis excluding studies at high risk of attrition bias | ||||||
| Tibolone compared with combined HT for postmenopausal women: adverse events | ||||||
| Population: postmenopausal women with or without vasomotor symptoms | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Combined HT | Tibolone | |||||
| Unscheduled bleeding (all doses) | 474 per 1000 | 224 per 1000 | OR 0.32 | 6438 | ⊕⊕⊝⊝ | |
| Endometrial cancer (all doses) | See comments | OR 1.47 | 3689 | ⊕⊝⊝⊝ | Events very rare in both groups. Total of 3 events: 2/1826 in tibolone group, 1/1863 in combined HT group | |
| Breast cancer; women without previous breast cancer (all doses) | 3 per 1000 | 6 per 1000 (3 to 13) | OR 1.69 | 4835 | ⊕⊝⊝⊝ | |
| Venous thromboembolic events (clinical evaluation; all doses) | 3 per 1000 | 1 per 1000 (0 to 6) | OR 0.44 | 4529 | ⊕⊝⊝⊝ | |
| Cardiovascular events (all doses) | 17 per 1000 | 10 per 1000 (4 to 27) | OR 0.63 | 3794 | ⊕⊝⊝⊝ | |
| Cerebrovascular event (all doses) | 1 per 1000 | 1 per 1000 (0 to 3) | OR 0.76 | 4562 | ⊕⊝⊝⊝ | |
| Mortality from any cause (tibolone 2.5 mg/d) | See comments | OR 3.05 | 970 | ⊕⊝⊝⊝ | Only 1 event (in tibolone group): 1/485 vs 0/485 | |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level for serious risk of bias: poor reporting of study methods and potential conflict of interest in some studies bDowngraded two levels for very serious risk of bias: poor reporting of study methods and potential conflict of interest in some studies cDowngraded one level for serious imprecision: low event rate. Findings compatible with meaningful benefit in one or both arms, or with no effect | ||||||
| Study | Comparator | Outcome measure | Information available | Notes | Results for meta‐analysis | SMD |
| Al‐Azzawi 1999 | HT | Presence of vasomotor symptoms, severity measured by Greene menopausal symptoms scale | 6 HRT and 9 tibolone patients were without symptoms at baseline. 67 HRT and 58 tibolone patients were free at month 3 | Contacted study authors, no reply | ||
| Baracat 2002 | HT | Total score: mean number of hot flushes per day multiplied by severity score | Means plotted as bar chart in Figure 1. Baseline, 11 for tibolone (n = 40), 12 for control (n = 45). At 3 months, 1.8 for tibolone and 1.5 for control. At 13 months, 0.2 for both | Would have to impute SDs – ‘no significant difference’ Unclear how to do this, given the available info Unable to find contact details | ||
| Benedeck‐Jaszmann 1987 | Placebo | 0 to 3 severity score | 12 months From Fig 1: Mean P: 1.6 T: 0.6 SD P: 1 T: 0.9 N P: 19 T: 24 (assuming 30 per arm to start, not explicitly stated) | Extracted from figure | Mean P: 1.6 T: 0.6 SD P: 1 T: 0.9 N P: 19 T: 24 | SMD: ‐1.0384784 SE: 0.3268612 |
| Bouchard 2012 | Placebo | Severity score | Calculate 12 week values P: 1.59 T: 1.16 Sample sizes of 150 (P) and 164 (T) Wk 12 | Use SD from sample size calc, which is in line with other studies | P: Mean 1.59 SD 0.9 N = 150 T: Mean 1.16 SD: 0.9 N = 164 | SMD: ‐0.4766282 SE: 0.1145686 |
| Egarter 1996 | HT | Severity of hot flushes (modified Kupperman Index) | Baseline mean C: 2.1 T: 2.2 6 months C: 0.4 T: 0.4 ‘N/S’ N = 34 (C) N = 62 (T) | Impute SD ‐ unclear how to Contacted study authors: no reply | ||
| Hammar 2007 | HT | Number of hot flushes | Week 48, baseline mean of both groups 6, follow‐up mean ≤ 1 Baseline SD C: 4.40 T: 4.37 N = 241 (C) N = 222 (T) | Use baseline SDs (these appear reasonable, given Landgren 2002) | C: mean 1, SD 4.40; N = 241 T: mean 1, SD 4.37 N = 222 | SMD: 0.00 SE: 0.09302624 |
| Hudita 2003 | Placebo (3 –arm study) | 5‐point severity scale for hot flushes | Week 24 P: 3 T: 1.25 mg: 0.2 T: 2.5mg: 0.1 N = 34 N = 45 N = 41 P < 0.01 for both compared with placebo | Split control group size between 2 arms Used P value to calculate SD Get implausible answers. Used known value instead (e.g. Hammar 1998) | Mean P: 3 T: 1.25 mg: 0.2 T: 2.5 mg: 0.1 N N = 34/2 = 17 N = 45 N = 41 SD P: 0.63 T: 1.25: 0.87 T: 2.5: 0.87 | 1.25 SMD: ‐3.4009511 SE: 0.4175209 2.5 SMD: ‐3.5375963 SE: 0.4371477 |
| Kokcu 2000 | HT | Occurrence of hot flushes | OR: 4.16 (0.75 to 22.9) | 2/19 have symptoms in C 12/19 have symptoms in T | SMD: 1.6236743 SE: 0.5369759 | |
| Landgren 2002 | Placebo (5‐arm study) | Frequency of hot flushes | Read means and SEs at 12 weeks from Figure 1 Mean P = 5.2 T 0.625 = 5 T 1.25 = 2.1 T 2.5 = 1.8 T 5.0 = 1.6 Standard error P = 0.37 T 0.625 = 0.37 T 1.25 = 0.40 T 2.5 = 0.43 T 5.0 = 0.37 Ns (calculated as all evaluable – dropouts ‐this assumes dropout occurred after 1st measurement at week 4) P = 113 T 0.625 = 129 T 1.25 = 124 T 2.5 = 139 T 5.0 = 136 | Read means and SEs from Figure 1 Calculated SDs using SEs and sample sizes Split placebo group size in 4 113/4 = 28.25 | Mean P = 5.2 T 0.625 = 5 T 1.25 = 2.1 T 2.5 = 1.8 T 5.0 = 1.6 SD P = 3.93 T 0.625 = 4.20 T 1.25 = 4.45 T 2.5 = 5.07 T 5.0 = 4.31 N (calculated as all evaluable – dropouts – this assumes dropout occurred after 1st measurement at week 4) P = 28.25 T 0.625 = 129 T 1.25 = 124 T 2.5 = 139 T 5.0 = 136 | 0.625 SMD: ‐0.04792794 SE: 0.20552850 1.25 SMD: ‐0.7077526 SE: 0.2102005 2.5 SMD: ‐0.6912512 SE: 0.2076033 5.0 SMD: ‐0.8437215 SE: 0.2097448 |
| Mendoza 2002 | HT | Flushes subscore of the Modified Kupperman Index, 0 to 2 score Number (%) reduced | Have number and percentage that improved in terms of vasomotor symptoms after 1 year Have 2 possible control groups – choose the best performing to give a conservative estimate 25/26 reduced in control group 27/29 reduced in T groups | Calculate odds ratio for reduced vasomotor symptoms. Turn this into an SMD for combination (27/2)/(25/1) = 0.54 SE log(OR) = Sqrt(1/27+1/2+1/25+1/1) = 1.26 | OR for improvement: OR = 0.54 SE(log(OR)) = 1.26 (so T worse) | SMD: 0.3734461 SE: 0.7610917 |
| Nappi 2006a | HT | Vasomotor symptoms (0 to 3 severity score) | At 6 months Means from Figure 4 C: 1.75 T: 1.5 P value for treatment term in ANOVA given as ‘P < 0.4’ N = 20 in both groups | Assume ANOVA P value is 0.4 and work out SDs as though this was a t‐test Gives SD of 0.657, assuming same in both groups | SMD: ‐0.3729492 SE: 0.3189649 | |
| Ross 1999 | HT | Greene Climacteric Scale subscore | Nothing usable. Only present 1 of 6 relevant comparisons because it is almost significant. Do not present 3 month score | |||
| Siseles 1995 | HT | Kupperman Index | No information given for vasomotor subscale | Have contacted study authors, no reply | ||
| Swanson 2006 | Placebo (3‐arm study) | Number of hot flushes per day | Median change from baseline at week 12 ‐5.5 P ‐9.7 T 2.5 ‐8.3 T 1.25 P < 0.001 for T 2.5 vs P P < 0.003 for T 125 vs P N P: 133 T 2.5: 125 T 1.25: 133 Actually, mean changes at week 12 and P values given in abstract T 2.5 vs P ‐10.14 vs ‐5.85, P < 0.001 T 1.25 vs P, week12 ‐8.32 P < 0.003 | Use reported values and calculate as for t‐tests. Split placebo group in half. Will have to impute SDs and final scores, as changes cannot be pooled with final scores if SMDs are used. For baseline, take median of values from Hammar 2007 and Landgren 2002 6,6,8,8,8,9,9.7 Mean 7.8. Too low – Figure 2 shows large changes. Say, 10 P: 10 ‐ 5.85 = 4.15 T 2.5: 10 ‐ 10 = 0 T 1.25: 10 ‐ 8.32 = 1.68 SDs too large when calculated from t‐test. Use values from Langren: P: 3.93 T 2.5: 5.07 T 1.25: 4.45 | Mean P: 10 ‐ 5.85 = 4.15 T 2.5: 10 ‐ 10 = 0 T 1.25: 10 ‐ 8.32 = 1.68 SD P: 3.93 T 2.5: 5.07 T 1.25: 4.45 N P: 66 T 2.5: 125 T 1.25: 133 | 1.25 SMD: ‐0.5741771133 SE: 0.1532927 2.5 SMD: ‐0.9661562 SE: 0.1599848 |
| Vieira 2009 | Placebo | Kupperman Index | Only overall Kupperman Index shown | Have contacted study authors, no reply | ||
| Volpe 1986 | Placebo HT | 0 to 9 score, with 0 = absent, 3 = mild, 6 = moderate, 9 = severe Unclear whether intermediate scores are possible | Can extract means for 24 weeks for tibolone arm, placebo arm and each of several HT arms, which have been partially combined, from Figure 1 in the paper | No real way to calculate SD from info in the paper, and the scale is different from those used in other studies (so not reasonable to use one from another study) | ||
| Wender 2004 | Placebo | Kupperman Index | Only overall Kupperman Index shown | Have contacted study authors, no reply |
| Study | Comparator | Outcome measure | Information available | Method used | Results for meta‐analysis | SMD |
| Hudita 2003 | Placebo (3‐arm study) | 0 to 4 scale | From figure Week 24 P: 2.6 T 1.25 mg: 1 T 2.5 mg: 0.9 N = 34/2 = 17 N = 45 N = 41 | Split control group size between 2 arms Use known value from other study for SD Use those from Nappi 2006a SD T: 0.89 HT: 0.89 | Mean P: 2.6 T 1.25 mg: 1 T 2.5 mg: 0.9 N N = 34/2 = 17 N = 45 N = 41 SD P: 0.89 T 1.25: 0.89 T 2.5: 0.89 | 1.25mg SMD: ‐1.7751711 SE: 0.3262804 2.5mg SMD: ‐1.8843965 SE: 0.3373802 |
| Kenemans 2009 | Placebo | Vaginal dryness as binary | P: 33/1558 T: 19/1575 | Convert OR to SMD | P: 33/1558 T: 19/1575 | |
| Swanson 2006 | Placebo (3‐arm study) | 0 to 3 score | Mean change from baseline at week 12 P: ‐0.2 T 2.5: ‐0.26 T 1.25: ‐0.39 N P: 133 T 2.5: 125 T 1.25: 133 | Split control group size between 2 arms Calculate final means using baseline and change – but no baseline values given Would also need to use SDs from another study | Cannot use | |
| Huber 2002 | HT | Vaginal dryness as binary | HT: 7/166 T: 6/158 | Convert OR to SMD | HT: 7/166 T: 6/158 | SMD: ‐0.06613757 SE: 0.34411866 |
| Kokcu 2000 | HT | Vaginal dryness as binary | HT: 0/21 T: 1/23 | Convert OR to SMD | HT: 0/21 T: 1/23 | SMD: 0.6382727 SE: 1.0064298 |
| Ziaei 2010 | HT and placebo | Vaginal dryness as binary Also, lubrication scores 1 to 5, higher is better – can reverse signs of mean differences | HT: 20/42 T: 33/47 P: 37/48 Mean HT: 4.93 T: 4.58 P: 3.65 SD HT: 1.95 T: 1.26 P: 1.81 | Use the continuous data Calculate and reverse sign, so that greater = increased vaginal dryness | HT: 20/42 T: 33/47 P: 37/48 | Using OR to SMD vs HT SMD: 0.5774306 0.2691251 vs placebo SMD ‐0.5904427 SE: 0.2096301 Using lubrication scores vs HT: SMD after switching sign: 0.2138954 SE: 0.2129393 vs placebo: SMD after switching sign: ‐0.1313959 SE: 0.2185150 |
| Nappi 2006a | HT | Vaginal dryness 0 to 3 score | From Figure 4, mean at 6 months Mean T: 0.7 HC: 0.6 SD: can read SE off Figure 4 and calculate SD N = 20 both groups | SD: can read SE off Figure 4 and calculate SD T: 0.89 HT: 0.89 | Mean T: 0.7 HC: 0.6 SD T: 0.89 HT: 0.89 N = 20 | SMD: 0.1101248 SE: 0.3164674 |
| Uygur 2005 | HT | 7‐point scale with ‐3 as worsened a lot and 3 as improved a lot | 6 months Mean (higher is better) HT: 0 T: 0.56 N HT: 34 T: 38 | P < 0.05 given. Assume P = 0.05 and calculate SD, assuming equal in 2 groups: Gives SD = 1.7 | Mean (higher is better) HT: 0 T: 0.56 N HT: 34 T: 38 Sd=1.7 for both | SMD after sign change: ‐0.3258676 0.2376236 |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Vasomotor symptoms Show forest plot | 7 | 1657 | Std. Mean Difference (Fixed, 95% CI) | ‐0.99 [‐1.10, ‐0.89] |
| 1.1 Tibolone 0.625 mg/d | 1 | 158 | Std. Mean Difference (Fixed, 95% CI) | ‐0.05 [‐0.46, 0.36] |
| 1.2 Tibolone 1.25 mg/day | 3 | 414 | Std. Mean Difference (Fixed, 95% CI) | ‐0.83 [‐1.06, ‐0.60] |
| 1.3 Tibolone 2.5 mg/day | 7 | 920 | Std. Mean Difference (Fixed, 95% CI) | ‐1.16 [‐1.30, ‐1.03] |
| 1.4 Tibolone 5 mg/day | 1 | 165 | Std. Mean Difference (Fixed, 95% CI) | ‐0.84 [‐1.25, ‐0.43] |
| 2 Unscheduled bleeding Show forest plot | 9 | 7814 | Odds Ratio (M‐H, Random, 95% CI) | 2.79 [2.10, 3.70] |
| 2.1 Tibolone, 2.5 mg/day | 8 | 4186 | Odds Ratio (M‐H, Random, 95% CI) | 2.58 [1.89, 3.52] |
| 2.2 Tibolone, 1.25 mg/day | 3 | 3628 | Odds Ratio (M‐H, Random, 95% CI) | 3.63 [2.37, 5.55] |
| 3 Endometrial cancer Show forest plot | 9 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Tibolone, all doses | 9 | 8504 | Odds Ratio (M‐H, Random, 95% CI) | 2.04 [0.79, 5.24] |
| 4 Breast cancer; women without previous breast cancer Show forest plot | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Tibolone, all doses | 4 | 5500 | Odds Ratio (M‐H, Random, 95% CI) | 0.52 [0.21, 1.25] |
| 5 Breast cancer; women with previous breast cancer Show forest plot | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Tibolone, 2.5 mg/day | 2 | 3165 | Odds Ratio (M‐H, Random, 95% CI) | 1.50 [1.21, 1.85] |
| 6 Venous thromboembolic events (clinical evaluation) Show forest plot | 5 | 9176 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.37, 1.97] |
| 6.1 Tibolone (all doses) | 5 | 9176 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.37, 1.97] |
| 7 Cardiovascular events Show forest plot | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Tibolone, all doses | 4 | 8401 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.84, 2.27] |
| 8 Cerebrovascular events; women's mean age over 60 years Show forest plot | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Tibolone (all doses) | 4 | 7930 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.99, 3.04] |
| 9 Mortality from any cause Show forest plot | 4 | 8242 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.79, 1.41] |
| 9.1 Tibolone, 2.5 mg/day | 3 | 3736 | Odds Ratio (M‐H, Random, 95% CI) | 0.94 [0.32, 2.73] |
| 9.2 Tibolone, 1.25 mg/day | 1 | 4506 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.54, 1.59] |
| 10 Insomnia Show forest plot | 3 | 3432 | Std. Mean Difference (Fixed, 95% CI) | ‐0.19 [‐0.38, ‐0.00] |
| 10.1 Tibolone, 2.5 mg/day | 3 | 3432 | Std. Mean Difference (Fixed, 95% CI) | ‐0.19 [‐0.38, ‐0.00] |
| 11 Vaginal dryness and painful sexual intercourse Show forest plot | 3 | 3348 | Std. Mean Difference (Fixed, 95% CI) | ‐0.66 [‐0.90, ‐0.43] |
| 11.1 Tibolone, 1.25mg/day | 1 | 62 | Std. Mean Difference (Fixed, 95% CI) | ‐1.78 [‐2.43, ‐1.13] |
| 11.2 Tibolone, 2.5 mg/day | 3 | 3286 | Std. Mean Difference (Fixed, 95% CI) | ‐0.49 [‐0.75, ‐0.24] |
| 12 Vaginal infections Show forest plot | 2 | 7639 | Odds Ratio (M‐H, Random, 95% CI) | 2.50 [1.24, 5.06] |
| 12.1 Tibolone, 2.5 mg/day | 1 | 3133 | Odds Ratio (M‐H, Random, 95% CI) | 1.73 [1.17, 2.55] |
| 12.2 Tibolone, 1.25 mg/day | 1 | 4506 | Odds Ratio (M‐H, Random, 95% CI) | 3.54 [2.61, 4.81] |
| 13 Urinary tract infections Show forest plot | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 Tibolone, 2.5 mg/day | 1 | 3133 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.46, 1.06] |
| 14 Endometrial hyperplasia Show forest plot | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 Tibolone, all doses | 4 | 4518 | Odds Ratio (M‐H, Random, 95% CI) | 1.20 [0.23, 6.25] |
| 15 Sensitivity Analysis ‐ Vasomotor symptoms without trials with high risk of attrition bias Show forest plot | 4 | Std. Mean Difference (Fixed, 95% CI) | ‐0.61 [‐0.73, ‐0.49] | |
| 15.1 Tibolone 0.625 mg/day | 1 | Std. Mean Difference (Fixed, 95% CI) | ‐0.05 [‐0.46, 0.36] | |
| 15.2 Tibolone 1.25 mg/day | 2 | Std. Mean Difference (Fixed, 95% CI) | ‐0.62 [‐0.86, ‐0.38] | |
| 15.3 Tibolone 2.5 mg/day | 4 | Std. Mean Difference (Fixed, 95% CI) | ‐0.65 [‐0.80, ‐0.50] | |
| 15.4 Tibolone 5 mg/day | 1 | Std. Mean Difference (Fixed, 95% CI) | ‐0.84 [‐1.25, ‐0.43] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Vasomotor symptoms Show forest plot | 2 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.35, 4.34] |
| 2 Insomnia Show forest plot | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Vaginal dryness and painful sexual intercourse Show forest plot | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 8.25] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Vasomotor symptoms Show forest plot | 9 | 1336 | Std. Mean Difference (Fixed, 95% CI) | 0.17 [0.06, 0.28] |
| 1.1 Tibolone, 2.5 mg/day | 9 | 1336 | Std. Mean Difference (Fixed, 95% CI) | 0.17 [0.06, 0.28] |
| 2 Unscheduled bleeding Show forest plot | 16 | 6438 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.24, 0.41] |
| 2.1 Tibolone, 2.5 mg/day | 16 | 4720 | Odds Ratio (M‐H, Random, 95% CI) | 0.34 [0.26, 0.45] |
| 2.2 Tibolone, 1.25 mg/day | 2 | 1718 | Odds Ratio (M‐H, Random, 95% CI) | 0.21 [0.16, 0.26] |
| 3 Endometrial cancer Show forest plot | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Tibolone, 2.5 mg/day | 5 | 3689 | Odds Ratio (M‐H, Random, 95% CI) | 1.47 [0.23, 9.33] |
| 4 Breast cancer; women without previous breast cancer Show forest plot | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Tibolone (all doses) | 5 | 4835 | Odds Ratio (M‐H, Random, 95% CI) | 1.69 [0.78, 3.67] |
| 5 Venous thromboembolic events (clinical evaluation) Show forest plot | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Tibolone (all doses) | 4 | 4529 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.09, 2.14] |
| 6 Cardiovascular events; all women's mean age below 60 years. No data available on different doses Show forest plot | 2 | 3794 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.24, 1.66] |
| 7 Cerebrovascular events; women's mean age below 60 years Show forest plot | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Tibolone (all doses) | 4 | 4562 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.16, 3.66] |
| 8 Mortality from any cause Show forest plot | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Tibolone, 2.5 mg/day | 2 | 970 | Odds Ratio (M‐H, Random, 95% CI) | 3.05 [0.12, 75.20] |
| 9 Endometrial hyperplasia Show forest plot | 5 | 2846 | Odds Ratio (M‐H, Random, 95% CI) | 0.35 [0.05, 2.21] |
| 9.1 Tibolone, 2.5 mg/day | 5 | 1549 | Odds Ratio (M‐H, Random, 95% CI) | 0.35 [0.04, 3.36] |
| 9.2 Tibolone, 1.25 mg/day | 1 | 1297 | Odds Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.48] |
| 10 Vaginal dryness and painful sexual intercourse Show forest plot | 7 | 1098 | Std. Mean Difference (Fixed, 95% CI) | 0.02 [‐0.12, 0.17] |
| 10.1 Tibolone, 2.5 mg/day | 7 | 1098 | Std. Mean Difference (Fixed, 95% CI) | 0.02 [‐0.12, 0.17] |
| 11 Sensitivity Analysis ‐ Vasomotor symptoms without trials with high risk of attrition bias Show forest plot | 4 | Std. Mean Difference (Fixed, 95% CI) | 0.25 [0.09, 0.41] | |
| 12 Sensitivity analysis ‐ vasomotor symptoms ‐ excluding studies with attrition bias and using nonvalidated scales Show forest plot | 3 | Std. Mean Difference (Fixed, 95% CI) | ‐0.03 [‐0.30, 0.23] | |
| 13 Vasomotor symptoms ‐ ordered by duration Show forest plot | 9 | Std. Mean Difference (Fixed, 95% CI) | 0.17 [0.06, 0.28] | |
| 13.1 Tibolone, 2.5 mg/day | 9 | Std. Mean Difference (Fixed, 95% CI) | 0.17 [0.06, 0.28] | |