Intervenciones para la fatiga en la neuropatía periférica
Resumen
Antecedentes
La sensación persistente de fatiga (o fatiga subjetiva), que se puede presentar en ausencia de factores fisiológicos, afecta a muchos pacientes con neuropatía periférica. Hay diversas intervenciones disponibles para la fatiga subjetiva, pero se conoce poco acerca de su eficacia o la probabilidad de algún efecto adverso en los pacientes con neuropatía periférica.
Objetivos
Evaluar los efectos de los fármacos y las intervenciones físicas, psicológicas o conductuales para la fatiga en adultos o niños con neuropatía periférica.
Métodos de búsqueda
El 5 noviembre 2013, se hicieron búsquedas en el registro especializado del Grupo Cochrane de Enfermedades Neuromusculares (Cochrane Neuromuscular Disease Group), CENTRAL, MEDLINE, EMBASE, CINAHL Plus, LILACS y AMED. También se buscó en las listas de referencias de todos los estudios identificados para la inclusión y revisiones pertinentes, y se estableció contacto con los autores de los estudios incluidos y expertos conocidos en el área para identificar datos adicionales publicados o no publicados. También se realizaron búsquedas de estudios en curso en registros de ensayos.
Criterios de selección
Se consideraron para inclusión los ensayos controlados aleatorizados (ECA) y cuasialeatorizados que compararan cualquier forma de intervención para el tratamiento de la fatiga en adultos con neuropatía periférica con placebo, ninguna intervención o una forma alternativa de intervención para la fatiga. Las intervenciones consideradas incluyeron fármacos, estimulación y modificación de la actividad física, ejercicios generales o específicos, estrategias compensatorias como estrategias con ortesis, relajación, orientación, cognitivas y educacionales.
Obtención y análisis de los datos
Dos autores de la revisión, de forma independiente, evaluaron el riesgo de sesgo y extrajeron los datos. Se contactó con los autores de los estudios para obtener información adicional. Se recopiló información sobre eventos adversos a partir de los ensayos incluidos.
Resultados principales
La revisión incluye tres ensayos, todos con bajo riesgo de sesgo, que reclutaron 530 pacientes con neuropatía periférica. Los efectos de la amantadina de un ensayo aleatorizado doble ciego controlado con placebo, cruzado, que comparó amantadina con placebo para el tratamiento de la fatiga en 80 pacientes con síndrome de Guillain‐Barré (SGB) fueron inciertos en la proporción de pacientes que lograron un resultado favorable a las seis semanas posteriores a la intervención (odds ratio [OR] 0,56; intervalo de confianza [IC] del 95%: 0,22 a 1,35; n = 74; p = 0,16). La calidad de esta evidencia se calificó como baja. Dos ensayos aleatorizados doble ciego controlados con placebo, de grupos paralelos, que compararon los efectos de dos dosis de ácido ascórbico con placebo para reducir la fatiga en adultos con enfermedad de Charcot‐Marie‐Tooth tipo 1A (CMT1A) mostró que los efectos del ácido ascórbico a cualquier dosis son probablemente pequeños (diferencia de medias estandarizada [DME] ‐0,12 (IC del 95%: ‐0,32 a 0,08; n = 404; p = 0,25]) para el cambio en la fatiga después de 12 a 24 meses (evidencia de calidad moderada). Ningún estudio de ácido ascórbico midió la fatiga a las cuatro a 12 semanas, que fue la medida de resultado primaria de esta revisión. No se informaron eventos adversos graves con la amantadina. En los ensayos de ácido ascórbico se informaron eventos adversos graves. Sin embargo, el riesgo de eventos adversos graves fue similar con ácido ascórbico y placebo.
Conclusiones de los autores
Un estudio pequeño poco preciso en pacientes con SGB mostró efectos inciertos de la amantadina sobre la fatiga. En dos estudios en pacientes con CMT1A hay evidencia de calidad moderada de que el ácido ascórbico tiene pocos efectos beneficiosos significativos sobre la fatiga. La información acerca de los efectos adversos fue limitada, aunque ambos tratamientos parecen ser bien tolerados y seguros en estas afecciones.
No hubo evidencia disponible de ECA para evaluar el efecto de otros fármacos u otras intervenciones para la fatiga en el SGB, la CMT1A u otras causas de neuropatía periférica. La relación entre costo y eficacia de las diferentes intervenciones también se debe considerar en los ensayos clínicos aleatorizados futuros.
PICOs
Resumen en términos sencillos
Tratamientos para la fatiga en la neuropatía periférica
Pregunta de la revisión
Evaluar los efectos de los tratamientos para la fatiga en pacientes con neuropatía periférica.
Antecedentes
La neuropatía periférica consiste en el daño de los nervios que se encuentran por fuera del cerebro y la médula espinal. Muchos pacientes con neuropatía periférica tienen sensación de cansancio grave (fatiga) que no está relacionada necesariamente con problemas físicos como la debilidad muscular. La evidencia en otras enfermedades a largo plazo en las que la fatiga es un problema indican que los fármacos y otras formas de tratamiento pueden ayudar. El objetivo de esta revisión fue evaluar el efecto de los fármacos y otros tratamientos como el ejercicio general o específico, la ortesis (dispositivos como férulas), la relajación, la orientación y las estrategias cognitivas, conductuales y educacionales sobre la sensación de fatiga en los pacientes con neuropatía periférica.
Características de los estudios
A partir de una amplia búsqueda, se identificaron tres ECA que cumplían los criterios de selección. Los ensayos involucraron a 530 adultos con neuropatía periférica. Los tratamientos fueron ácido ascórbico (vitamina C) en dos ensayos en pacientes con enfermedad de Charcot‐Marie‐Tooth, y amantadina en el síndrome de Guillain‐Barré en el tercer ensayo. La enfermedad de Charcot‐Marie‐Tooth es una enfermedad hereditaria de los nervios y el síndrome de Guillain‐Barré es una afección en la que hay inflamación de los nervios periféricos. Se seleccionaron las medidas de fatiga a las cuatro a 12 semanas como las medidas preferidas de los efectos del tratamiento. El ensayo de amantadina, pero no los ensayos de ácido ascórbico, proporcionaron datos en este punto temporal; los ensayos de ácido ascórbico proporcionaron datos más de 12 meses después del comienzo de la intervención.
Resultados clave y calidad de la evidencia
No hubo evidencia suficiente para determinar los efectos de la amantadina comparada con un tratamiento inactivo (placebo) para la fatiga, y no hubo efectos no deseados importantes. Se encontró evidencia de que el ácido ascórbico probablemente tiene un efecto beneficioso poco significativo en la fatiga. No se identificaron efectos no deseados importantes, pero los ensayos fueron pequeños. La calidad de esta evidencia se evaluó como moderada porque los resultados fueron imprecisos, que significa que no descartan la posibilidad de que los fármacos puedan tener un efecto. No se encontró evidencia de ECA sobre otros fármacos o tratamientos para la fatiga en la neuropatía periférica.
No hay evidencia suficiente para determinar el efecto de la amantadina para la fatiga en el SGB y del ácido ascórbico para la fatiga en la enfermedad CMT1A. Se necesitan más estudios de alta calidad que proporcionen evidencia sobre las cuales basar el tratamiento de la sensación de fatiga en la neuropatía periférica. La relación entre costo y eficacia de las diferentes intervenciones también se debe considerar en los ECA futuros.
La evidencia de esta revisión está actualizada hasta noviembre 2013.
Authors' conclusions
Summary of findings
| Amantadine versus placebo for fatigue in Guillain‐Barré syndrome | ||||||
| Patient or population: people with fatigue in Guillain‐Barré syndrome | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Amantadine | |||||
| Reduction in fatigue of at least 1‐point on the Fatigue Severity Scale between 4 to 12 weeks after commencement of intervention | 257 per 1000 | 162 per 1000 | OR 0.56 | 74 | ⊕⊕⊝⊝ | ‐ |
| Change in fatigue 12 or more weeks after commencement of intervention (not measured) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in activity limitation 12 or more weeks after commencement of intervention (not measured) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in participation 12 or more weeks after commencement of intervention using the | The mean change in participation 12 or more weeks after commencement of intervention using the control group was | The mean change in participation 12 or more weeks after commencement of intervention using the intervention group was | 74 | ⊕⊕⊝⊝ | ‐ | |
| Health related quality of life after 12 or more weeks or more after commencement of intervention (not reported) | ‐ | ‐ | ‐ | ‐ | ‐ | EHQ and SF‐36 measured but data not fully reported |
| Serious adverse events | See comment | See comment | Not estimable | 80 | See comment | 0 participants had serious adverse events in this study |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Evidence downgraded two levels due to imprecision (small sample size and wide CI that crosses the line of no effect). | ||||||
| Ascorbic acid versus placebo for fatigue in peripheral neuropathy | ||||||
| Patient or population: people with fatigue in peripheral neuropathy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Ascorbic acid | |||||
| Reduction in fatigue of at least 1 point on the Fatigue Severity Scale (FSS) between 4 to 12 weeks after commencement of intervention (not measured) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in fatigue 12 or more weeks after commencement of intervention | ‐ | The mean change in fatigue 12 or more weeks after commencement of intervention in the intervention groups was | ‐ | 404 | ⊕⊕⊕⊝ | ‐ |
| Change in activity limitation (ODSS/ONLS) 12 or more weeks after commencement of intervention | See comment | See comment | Not estimable | 406 | ‐ | Data available were not suitable for meta‐analysis |
| Change in 10 metre timed walk 12 or more weeks after commencement of intervention | The mean changes in 10 metre timed walk 12 or more weeks after commencement of intervention in the control groups werea decrease of 0.40 seconds and an increase of 0.76 seconds2 | The mean change in 10 metre timed walk 12 or more weeks after commencement of intervention in the intervention groups was a decrease of | 406 | ⊕⊕⊕⊝ | ‐ | |
| Change in speed of completion of 9‐hole peg test (in seconds) 12 or more weeks after commencement of intervention | The mean change in speed of completion of the 9‐hole peg test was an increase of 0.85 seconds in the control group | The mean change in speed of completion of the 9‐hole peg test was a decrease of 0.74 seconds (1.60 faster to 0.12 slower) | Not estimable | 224 | ⊕⊕⊕⊝ | ‐ |
| Health‐related quality of life 12 or more weeks after commencement of the intervention Change in SF‐36 physical function score | ‐ | The mean health‐related quality of life 12 or more weeks after commencement of the intervention (change in SF‐36 physical function score) in the intervention groups was | ‐ | 400 | ⊕⊕⊕⊝ | ‐ |
| Change in participation 12 or more weeks after commencement of intervention (not measured) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Health‐related quality of life 12 or more weeks after commencement of the intervention Change in mental function score of SF‐36 | The mean change in health‐related quality of life 12 or more weeks after commencement of the intervention (change in mental function score of SF‐36) in the control group was 1.2 points | The mean health‐related quality of life 12 or more weeks after commencement of the intervention (change in mental function score of SF‐36) in the intervention groups was | ‐ | 123 | ⊕⊕⊕⊝ | ‐ |
| Serious adverse events | 108 per 1000 | 76 per 1000 | RR 0.70 | 450 | ⊕⊕⊕⊕ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Only one or two studies included. | ||||||
Background
Description of the condition
Peripheral neuropathies collectively affect about 2.4% of the population and may be either genetic or acquired, and either acute or chronic in nature (Martyn 1998). The pathological process affects peripheral nerves, resulting in neurological damage to either the axon (ie, degeneration of the central nerve fibre), the myelin (ie, demyelination or destruction of the insulating sheath of the nerve), or a combination of both. Recovery may be by remyelination, through which improvements in function can be rapid, and in some people there is almost complete recovery. However, regeneration of damaged axons usually takes many months or years and recovery, if present, may be incomplete (Tamura 2007).
Common symptoms of peripheral neuropathy include numbness, diminished or altered sensation (pins and needles), muscle weakness, and autonomic dysfunction. People may also experience fatigue, pain, psychological dysfunction and poor social adjustment (Lennon 1993; Pfeiffer 2001). Even when neurological function improves, residual symptoms often persist and fatigue is frequently an ongoing problem (Merkies 1999).
The experience of fatigue (or subjective fatigue) has been described as a "an overwhelming sense of tiredness, lack of energy and feeling of exhaustion" that is "not relieved by rest" and is a common sequel of chronic conditions (Bleijenberg 2003; Karlsen 1999; Krupp 2003). Physiological fatigue, or "the loss of voluntary force‐producing capacity during exercise" (Bigland‐Ritchie 1978) can contribute to subjective fatigue, but people can experience subjective fatigue in the absence of physiological factors and physiological fatigue does not necessarily result in subjective fatigue.
Physical factors such as residual muscle weakness mean that people with peripheral neuropathy have to work harder during everyday activities than healthy individuals, which will feel more of an effort. Axonal degeneration prevents nerve impulses from being conducted along the peripheral nerve and demyelination can lead to conduction block that can be exacerbated by muscle activity (Cappelen‐Smith 2000), leading to fatigue (Kaji 2000). Although there is no direct problem with the muscles themselves, it has been shown that people with peripheral neuropathy may be unable to voluntarily activate their muscles fully, possibly because of dysfunction within the central nervous system (Garssen 2007; Schillings 2003). This form of physiological fatigue is known as central fatigue and could possibly be due to reduced drive from nerve cells in the brain (cortical neurons) or to the effect of alterations in the number of surviving peripheral nerve axons and an increase in the number of muscle fibres they innervate (Martinez‐Figuerora 1977; Sanders 1996). In addition, psychological factors such as lack of motivation or low mood can also affect voluntary activation of muscles (Gandevia 2001; Kent‐Braun 1993). Fear of precipitating fatigue may also contribute and lead to an avoidance of physical activity and physical deconditioning, thus further exacerbating fatigue (Moss‐Morris 2005). Environmental factors may also precipitate fatigue, since many people with peripheral neuropathies have to utilise compensatory strategies such as orthoses or walking aids as a result of deformities or sensory deficits. Therefore, the experience of fatigue may be clearly related to known underlying factors or, more commonly, be only partly explained by these. Finally, measurement of the level of fatigue is difficult both because the subjective experience of fatigue varies between individuals and because measures of different types of fatigue are numerous.
Description of the intervention
The evidence about the efficacy of interventions for fatigue in peripheral neuropathy and other chronic conditions is limited and unclear. Interventions aim to address physical, environmental and psychological factors contributing to fatigue, and include drugs, pacing and grading of physical activity, general or specific exercise, orthotics, relaxation, counselling, cognitive behavioural strategies and others.
How the intervention might work
The presumed mode of action of drug interventions such as amantadine for generalised fatigue is unknown (Pucci 2009); one randomised controlled trial (RCT) showed no effect in inflammatory peripheral neuropathy (Garssen 2006). It is likely that factors contributing to subjective fatigue in individuals with peripheral neuropathy must be clearly understood in order to target interventions effectively. For example, where fatigue is due to weakness, strengthening exercise may be beneficial, but where altered mood or well‐being is a factor then drug, cognitive or behavioural interventions may be more relevant. It is likely that multidimensional interventions that address individuals' self perception and health beliefs combined with strategies to increase levels of physical activity or participation in regular exercise or both may also be helpful.
Why it is important to do this review
Fatigue is a frequent and often severe problem that affects everyday activities and quality of life for people with peripheral neuropathy. Although there have been some RCTs of drug and non‐drug interventions for fatigue in peripheral neuropathy, we know of no systematic review.
Objectives
To assess the effects of drugs and physical, psychological or behavioural interventions for fatigue in adults or children with peripheral neuropathy.
Methods
Criteria for considering studies for this review
Types of studies
We included all randomised controlled trials (RCTs) or quasi‐RCTs of any drug or non‐drug intervention to treat fatigue associated with peripheral neuropathy, compared with placebo, no treatment or other drug or non‐drug interventions for fatigue. Quasi‐RCTs are those in which randomisation was intended but they use methodology that may be biased, such as alternation, or use of case record numbers or date of birth.
Types of participants
We considered for inclusion in the review all trials in which participants (adults or children) had fatigue associated with a diagnosis of peripheral polyneuropathy, including sensory, motor and combined sensory and motor neuropathies. We did not consider trials including mostly people with focal disease such as local entrapment neuropathies with pain as the primary presenting feature (for example, cervical radiculopathy, carpal tunnel syndrome, brachial plexus neuritis, etc), and poliomyelitis. We accepted the diagnosis of peripheral neuropathy offered by the study authors, provided that it stipulated the presence of clinical impairment characteristic of peripheral neuropathy. We did not include diagnoses dependent on symptoms suggestive of neuropathy alone or neurophysiological abnormalities in the absence of clinical signs.
Types of interventions
We considered trials including any form of intervention for fatigue management, such as drugs, pacing and grading of physical activity, general or specific exercise, compensatory strategies such as orthotics, relaxation, counselling, cognitive and educational strategies and others, compared with either placebo, no intervention or an alternative form of intervention for fatigue.
Types of outcome measures
Primary outcomes
Our primary outcome was fatigue severity and symptoms as measured by any validated scale evaluated at least four weeks and less than 12 weeks after commencement of the intervention. Where studies used more than one scale, we used the preferred ranking of commonly‐used scales shown below to identify the scale to be used in the primary outcome analysis. Instruments that we considered appropriate included: Fatigue Severity Scale (FSS) (Krupp 1989), Chalder Fatigue Scale (Chalder 1993), Fatigue Impact Scale or daily‐FIS (D‐FIS) (Fisk 1994; Fisk 2002), Visual Analogue Scale of Fatigue (VAS‐F) (Lee 1991), and Piper Fatigue Scale (PFS) (Piper 1998).
Secondary outcomes
1. Fatigue after 12 or more weeks.
2. Activity limitations after 12 or more weeks.
3. Participation restrictions after 12 or more weeks.
4. Health‐related quality of life after 12 or more weeks.
5. Adverse events: any adverse events, adverse events which lead to discontinuation of treatment and serious adverse events, which are those which are fatal, life‐threatening, or require prolonged hospitalisation.
Search methods for identification of studies
Electronic searches
On 5 November 2013, we searched the Cochrane Neuromuscular Disease Group Specialized Register, the Cochrane Central Register of Controlled Trials (CENTRAL) (2013, Issue 10), MEDLINE (January 1966 to October 2013), EMBASE (January 1980 to October 2013), LILACS (January 1982 to October 2013), CINAHL Plus (January 1937 to October 2013) and AMED (January 1985 to October 2013). The detailed search strategies are in the appendices: Appendix 1 (MEDLINE), Appendix 2 (EMBASE), Appendix 3 (CINAHL Plus), Appendix 4 (CENTRAL), Appendix 5 (AMED), Appendix 6 (LILACS) and Appendix 7 (the Cochrane Neuromuscular Disease Group Specialized Register).
On 12 June 2014 we searched the Current Controlled Trials register (www.controlled‐trials.com), ClinicalTrials.gov (www.clinicaltrials.gov) and the WHO International Clinical Trials Registry Platform (ICTRP) (www.who.int/ictrp/en/), using the search terms 'fatigue' and 'peripheral neuropathy'.
Searching other resources
We reviewed the bibliographies of the randomised trials identified, contacted the trial authors and known experts in the field, and approached pharmaceutical companies to identify additional published or unpublished data.
Data collection and analysis
Selection of studies
Two review authors (CMW, RCS) independently examined the titles and abstracts identified by the search. There were no language restrictions. We obtained English language translations where appropriate. We retrieved all trials that were relevant and reviewed full‐text publications. We examined retrieved publications against the selection criteria and we resolved any disagreement by consensus.We did not have to resort to arbitration by a third author (MPG). If trials had had a heterogenous sample of disorders, we would have only included them if data from participants with peripheral neuropathy could be isolated or if at least 75% of the participants had peripheral neuropathy.
Data extraction and management
For each retrieved publication, two review authors (CW and RCS) independently extracted the relevant data using standardised forms. We resolved any disagreements by discussion. We gathered data on:
-
eligibility criteria;
-
interventions;
-
details of participants;
-
assignment to groups;
-
outcome measures;
-
time at which outcomes are measured;
-
funding;
-
declarations of interest;
-
sample size; and
-
statistical analysis.
Assessment of risk of bias in included studies
Two out of three review authors (CW, RS and MG) independently assessed all included studies for risk of bias. We graded the items according to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a) and presented our assessments in a 'Risk of bias' table. We assessed trials for random sequence generation, allocation concealment, blinding (participants, personnel and outcome assessors), incomplete outcome data, selective outcome reporting and other potential sources of bias.
We then made a judgement on each of these criteria relating to the risk of bias, of 'low risk of bias', 'high risk of bias' or 'unclear risk of bias'.
Measures of treatment effect
Continuous data
For studies using the same outcome measures, we summarised continuous data using mean differences (MDs), sometimes called 'weighted mean differences', with a corresponding 95% confidence interval (CI). When different studies measured the same outcome in different ways we analysed results using standardised mean differences (SMDs) with a 95% CI.
Dichotomous data
For studies using similar outcomes with dichotomous data we calculated a pooled estimate of the risk ratio (RR), with a 95% CI.
Unit of analysis issues
Where there were multiple intervention groups within a trial, we made pairwise comparisons of similar interventions or active components versus no treatment, placebo, or another intervention. We planned to analyse cross‐over trials with continuous outcome measures using generalised inverse variance analysis (GIV) (DerSimonian 1986), where the estimated difference in the mean treatment effects and their standard errors were available for pooling with equivalent results from parallel‐group studies. If this information had not been available then we would have pooled data from the first phase only, where possible, with parallel design studies. For cross‐over studies with dichotomous outcomes we would have pooled results using odds ratios (ORs) and confidence intervals from paired analyses where available (Elbourne 2002). Only one cross‐over trial was included for any comparison, so no decisions about pooling data were necessary. We would have reported trials with unique designs or outcomes without a meta‐analysis. We excluded studies that were not RCTs from the analyses but we comment on them in the Discussion.
Dealing with missing data
For all outcomes, where continuous data were presented as mean change with 95% CIs, the standard deviations (SDs) were calculated for input into analyses and forest plots assuming a normal distribution due to a moderate sample size, using the equation SD = SQRT of number of participants X (upper limit of CI to lower limit of CI) / 3.92. as recommended in Section 7.7.3.2 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). Where overall disability sum score (ODSS) data were presented as median and interquartile ranges, we did not input any data for analysis. Where participant dropout led to missing data we contacted trial authors if an intention‐to‐treat analysis had not been performed. We also contacted authors where there were missing data for review outcomes, and additional data were provided for the secondary outcome of participation in one study (Garssen 2006).
Assessment of heterogeneity
We undertook meta‐analysis when studies investigated similar interventions, used similar outcome measures and included groups of participants who were clinically homogeneous. Where studies were heterogeneous for some outcomes we undertook a narrative review.
If studies were similar in intervention, outcome measurement and participants, we assessed possible inconsistency across studies using the I² statistic (Higgins 2003; Higgins 2011a). If studies had been heterogeneous (Q statistic = 0.1 and I² statistic of 25% or greater), the review authors would have considered conducting subgroup analysis only. If we had considered the primary studies to be heterogeneous even within subgroups, we would have undertaken a narrative approach rather than a meta‐analysis.
Assessment of reporting biases
We did not use funnel plots to investigate associations between effect size and study precision in terms of sample size, because of the small number of studies included in the review. If we had included funnel plots, we would have explored the relationships observed to investigate differences between studies with large and small samples, or systematic biases, such as publication bias.
Data synthesis
For studies with a similar type of intervention or a similar active component, we performed a meta‐analysis to calculate a treatment effect across trials with a fixed‐effect model, as heterogeneity was not demonstrated. Dichotomous outcome results were available from only one study so we did not present pooled RRs. Where it was not possible to perform a meta‐analysis we provided descriptive summaries of the results from each trial.
'Summary of findings' table
We created 'Summary of findings' tables using the following outcomes:
-
Fatigue severity and symptoms between four and 12 weeks after commencement of the intervention.
-
Fatigue after 12 or more weeks.
-
Activity limitations after 12 or more weeks.
-
Participation restrictions after 12 or more weeks.
-
Health‐related quality of life after 12 or more weeks.
-
Serious adverse events.
We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of a body of evidence (studies that contribute data) for the prespecified outcomes. We used methods and recommendations described in Chapters 11 and 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011a; Schünemann 2011b). We used GRADEpro software (GRADEpro 2008). We explained decisions to down‐ or up‐grade the quality of studies using footnotes and we made comments to aid reader's understanding of the review where necessary.
Subgroup analysis and investigation of heterogeneity
Insufficient data available. See Appendix 8 for methods planned but not implemented.
Sensitivity analysis
Insufficient data available. See Appendix 8 for methods planned but not implemented.
This review has a protocol (White 2009). We listed any deviations from the protocol in Differences between protocol and review.
Results
Description of studies
See Characteristics of included studies and Characteristics of excluded studies for details.
The electronic and manual searches identified a total of 1386 titles and abstracts. After removing duplicates and excluding abstracts where studies were clearly not eligible for inclusion, only six full publications, four protocols and two abstracts were relevant to the review. After reading the full text of the six published articles we excluded three: two were not randomised controlled trials (RCTs) (Carter 2006; Enderlin 2008) and one was an RCT of ascorbic acid for children with CMT1A in which fatigue was not an outcome (Burns 2009). We included one abstract of conference proceedings, regarding an RCT of a new low‐dose combination of three already approved drugs for CMT1A, as an ongoing trial, as the analyses are ongoing (Attarian 2013). We will include this study in future updates of this review if appropriate. Two of the other five ongoing or unpublished studies were RCTs of ascorbic acid in CMT1, and we contacted the authors for further information (NCT00484510; Verhamme 2009). However, in these ongoing studies, fatigue was not included as an outcome and we excluded them. One randomised trial of coenzyme Q10 for CMT (NCT00541164), with changes in weakness, fatigue and pain as the primary outcome, is completed but appears to be unpublished. NCT02121678 is not yet recruiting, but will study resistance training for chronic inflammatory demyelinating neuropathy (CIDP) with the fatigue severity scale (FSS) as a planned secondary outcome. We identified one study, Ramdharry 2011, from an abstract in the Cochrane Neuromuscular Disease Group Specialized Register. The study appears to be ongoing and the authors provided no data when contacted. We will include it if eligible once the full publication is available. See Characteristics of ongoing studies.
The study selection process therefore resulted in only three trials finally meeting the criteria for inclusion (Garssen 2006; Micallef 2009; Pareyson 2011).
Study design and participants
We include three studies evaluating the efficacy of two drugs for fatigue in two different populations (Garssen 2006; Micallef 2009; Pareyson 2011).
The first study (Garssen 2006) was a randomised, double‐blind, placebo‐controlled, cross‐over trial of amantadine for severe fatigue in 80 neurologically well‐recovered participants who had had Guillain‐Barré syndrome (GBS). Randomisation was in blocks of six with half the study participants receiving amantadine first and the remainder receiving placebo first. Interventions were followed by a two‐week washout period. Assessment of outcomes occurred on six occasions: 1: at baseline, 2: following two weeks of no intervention, 3: after six weeks of intervention 1, 4: after a two‐week washout period, 5: after six weeks of intervention 2, and finally 6: after a further two‐week washout period. Three randomised participants were withdrawn from the study in the pre‐cross‐over phase. One participant was in the placebo group and withdrew because of concern over potential side effects of amantadine when she became pregnant. The two other participants were in the amantadine treatment group; one withdrew because of hospital admission with acute cholangitis and the second participant developed severe complaints of dizziness. A further three participants were excluded from the analysis due to incorrect completion of the FSS. Therefore, 74 participants with GBS were included in the reporting of outcomes. All participants had been diagnosed with GBS between six months and 15 years previously and had no apparent changes in GBS disability score for the three months prior to entering the study. All could walk at least 10 metres with or without a walking aid (GBS disability score < 3).
The two remaining studies (Micallef 2009; Pareyson 2011) were parallel‐group, randomised, double‐blind, placebo‐controlled trials of the effect of ascorbic acid in a total of 450 adults with Charcot‐Marie‐Tooth disease type 1A (CMT1A). In one, two doses (either 1 g or 3 g daily) of ascorbic acid or placebo were used in the treatment of 179 participants (Micallef 2009). Confirmation of diagnosis was by clinical examination and genotyping with duplication in 17p11.2 and at least one motor sign or symptom (gait disorder, distal amyotrophy, foot deformation or distal weakness). Block randomisation into three groups with matching for study site and sex was followed by 12 months of treatment with either 1 g or 3 g of ascorbic acid or placebo; outcomes were assessed at baseline and after 12 months of intervention. A total of 16 participants withdrew from the study after randomisation due to adverse events (six), withdrawal of consent (six), loss to follow‐up (two), non‐compliance (one) or missing baseline data (one).
Therefore, 163 participants completed the study but analysis was of all 179 participants, on an intention‐to‐treat basis.
In the second multicentre study (Pareyson 2011), the efficacy of chronic administration, for 24 months, of either 1.5 g/day ascorbic acid or placebo in the treatment of 271 participants (eligible for analysis) with Charcot‐Marie‐Tooth disease 1A was evaluated. Participants had a clinical diagnosis of Charcot‐Marie‐Tooth disease 1A with genetic confirmation of 17p11.2‐p12 duplication and a CMT neuropathy score of between 1 (excluding the electrophysiological component) and 35 (including the electrophysiological component). Block randomisation of 277 participants into groups of four stratified by disease severity and centre was followed by 24 months of treatment with either ascorbic acid or placebo, and outcomes were assessed at six‐monthly intervals from commencement of intervention until 24 months. Six participants did not receive the allocated intervention after randomisation, and are dropped from the denominators. A total of 22 participants withdrew from the intervention after commencement. Reasons for withdrawal were adverse events (13), withdrawal of consent (six), moved away from study area (two) or for personal reasons (one). Whilst the primary study outcome was based on all randomised patients who received at least one dose of the study drug using imputation for missing data the secondary analyses were conducted without imputation of missing data.
Interventions
The study of amantadine in Guillain‐Barré syndrome was a cross‐over trial in which amantadine was compared with placebo at a dose of one tablet (100 mg) daily for one week followed by two tablets daily for the remaining five weeks of each intervention period. A washout period of two weeks was incorporated before the participants shifted to the other treatment arm (amantadine or placebo) (Garssen 2006).
The studies of ascorbic acid in Charcot‐Marie‐Tooth disease 1A were a parallel‐group study comparing 1 g or 3 g of ascorbic acid in a single administration of three capsules with three placebo capsules for 12 months (Micallef 2009) and a parallel‐group study comparing 1.5 g of ascorbic acid in two daily doses with placebo tablets for 24 months (Pareyson 2011).
Outcomes
In participants with severe fatigue due to Guillain‐Barré syndrome (defined as Fatigue Severity Scale (FSS) 5.0 or more; FSS range 1 to 7), Garssen 2006 used a reduction of at least one point on the FSS as a primary outcome measure of fatigue. Outcomes for FSS were reported for both phases of the cross‐over study. Secondary outcome measures in the major domains of the International Classification of Functioning, Disability and Health (ICF) were: for structure and function, anxiety and depression using the Hospital Anxiety and Depression Scale (HADS); for activity, impact of fatigue using the Fatigue Impact Scale (FIS); and for participation, Rotterdam Handicap Scale (RHS), quality of life using the Medical Outcomes Short Form‐36 (SF‐36) Health Survey and the Euroquol Health Questionnaire (EQHQ); and adverse events.
In the studies of ascorbic acid in participants with Charcot‐Marie‐Tooth disease 1A, the primary outcome was CMT neuropathy score (CMTNS). Fatigue was assessed as a secondary outcome using the change in Visual Analogue Scale ‐ Fatigue (VAS‐F), either 12 months (Micallef 2009) or 24 months (Pareyson 2011) after commencement of the intervention. Secondary outcomes of activity limitation using the Overall Disability Sum Score (ODSS) (Micallef 2009) or Overall Neuropathy Limitations Scale (ONLS) (Pareyson 2011), 10 metre walk test (10MWT) Pareyson 2011 and 9 hole peg test (9HPT) (Pareyson 2011) were reported, as well as measures of participation and quality of life using the SF‐36 and adverse events in both studies.
Risk of bias in included studies
See Characteristics of included studies for details and Figure 1.

Methodological quality summary: review authors' judgements about each methodological quality item for each included study. Key: green (+) = low risk of bias; yellow (?) = unclear risk of bias; red (‐) = high risk of bias.
Garssen 2006 conducted block randomisation in blocks of six, although allocation was concealed from researchers; we therefore considered the study to be at low risk of bias for these criteria. The trial was a double‐blind, placebo‐controlled study, with 80 participants initially enrolled. Although the final analysis was based on 74 participants as six were withdrawn, there was adequate reporting of the loss to follow‐up. Sample size was calculated using estimates of proportion from unpublished data obtained from a pilot study, indicating that 36 participants per group were required for 90% power at a two‐sided 5% alpha, so that with 77 participants completing the trial it was sufficiently powered to detect a difference. The washout period of two weeks between cross‐over intervention phases seems sufficient to avoid a carry‐over effect of amantadine, and is comparable with other randomised controlled cross‐over designs of efficacy for amantadine in other populations (Cohen 1989; Pucci 2009; Rosenberg 1988). The authors reported data for mean (SD) improvement in FSS scores for both amantadine and placebo treatment in both the amantadine‐placebo (0.46 (1.29), P = 0.078; 95% CI ‐0.92 to 0.05) and placebo‐amantadine sequence (0.08 (1.44), P = 0.28; 95% CI ‐1.24 to 0.44) groups and t‐test analysis for period effects and carry‐over effects were not significant. There was some evidence of selective reporting of secondary outcomes, as only P values were reported. Overall we considered the study to be at low risk of bias.
In Micallef 2009, sequence generation was conducted by an independent contract research organisation who were also responsible for concealed allocation of randomisation into blocks of 12 that were stratified for sex and study site (three sites). The trial was double‐blind with the administration of visually identical hard gelatin capsules for all groups. Analysis was on an intention‐to‐treat basis with 179 participants randomised and analysed despite 15 withdrawals prior to follow‐up (four from the placebo group, eight from the 1 g ascorbic acid group and three from the 3 g ascorbic acid group); one participant from the placebo group had missing baseline data for the study's primary outcome. All study outcomes were adequately reported. Whilst authors stated that the data were analysed on an intention‐to‐treat basis there was no indication of how the investigators dealt with the missing data from 16 participants. Our overall summary assessment was that the study was at low risk of bias.
In the third study (Pareyson 2011), an independent randomisation unit was responsible for sequence generation and group allocation stratified for disease severity and treatment centre in blocks of four. This, combined with the use of placebo tablets of identical appearance, taste and smell, resulted in blinding of participants, researchers and treating physicians. Six participants randomised to the intervention (four participants) and placebo (two participants) groups withdrew consent before receiving the allocated intervention and were excluded from analysis. Although the investigators recorded outcome data at six‐monthly intervals for 24 months, the authors stated that their main analysis would be at 24 months, and reported the primary outcome at only 12 and 24 months and the secondary outcomes only at 24 months. Overall we considered this study to be at low risk of bias.
Effects of interventions
See: Summary of findings for the main comparison Amantadine versus placebo for fatigue in Guillain‐Barré syndrome; Summary of findings 2 Ascorbic acid versus placebo for fatigue in peripheral neuropathy
Amantadine versus placebo in Guillain‐Barré syndrome
Effect estimates, where data were available, were obtained from a single trial, so we downgraded the quality of evidence for all outcomes for this reason (summary of findings Table for the main comparison).
Primary outcome
Fatigue between four and 12 weeks after commencement of intervention
Garssen 2006 assessed baseline measures at visit 1 and at visit 2, prior to randomisation. In the 74 participants for whom full outcome data were reported, there was evidence of a reduction in fatigue between these baseline visits, which was stated by the authors to approach significance (P = 0.05 Wilcoxon signed rank test), although complete group data were not presented. The magnitude of fatigue reduction before the start of any intervention was such that although the median FSS score for the 74 participants was 5.9 at visit 2, seven (9%) of the participants had an FSS score lower than the inclusion criterion of FSS 5 for severe fatigue.
Thirty‐six participants received amantadine followed by placebo and 38 received placebo followed by amantadine. Favourable outcome reporting was available for both phases of each treatment sequence, representing evaluation of the primary outcome six weeks after the commencement of intervention. Results were presented as the mean difference (MD) in reduction in FSS scores between amantadine and placebo, and as an odds ratio (OR) of a favourable outcome (defined as a reduction of more than 1 in FSS score). The overall MD in FSS score between amantadine and placebo phases was ‐0.45 (95% CI ‐0.94 to 0.04; t = ‐1.80; df = 73; P = 0.076). When the participants responding favourably to one treatment only in both the group receiving amantadine followed by placebo (amantadine‐placebo group) and those receiving placebo followed by amantadine (placebo‐amantadine group) were combined, the number with a more favourable outcome following amantadine was 9 of 25 (36%) participants with an OR of 0.56 (95% CI 0.22 to 1.35, P = 0.16) using the McNemar test.
Secondary outcomes
All stated secondary outcomes in Garssen 2006 were only reported at six weeks after commencement of the intervention and therefore not at the stated secondary outcome point of 12 or more weeks after commencement of intervention for this review. In addition full data for the study secondary outcomes were not available, as only P values were provided (Garssen 2006). The data indicated that there was no significant improvement in: fatigue impact using the fatigue impact scale (FIS) (P = 0.77); participation using the Rotterdam Handicap Scale (RHS) (P = 0.14) and Euroquol Health Questionnaire (EQHQ) (P = 0.21) at six weeks. The authors reported that quality of life using the SF‐36 demonstrated a significant improvement in physical role functioning (P = 0.008) and mental health perception (P = 0.008), although only in the placebo group (Garssen 2006). We contacted the authors for further information about secondary outcomes; the RHS scores for participation at six weeks from the original data were provided by the trial author (Garssen 2006). The data were analysed using a paired two‐sample t‐test and showed that the MD between the change in participation with placebo versus the change with amantadine between four to 12 weeks after commencement of intervention was 0.31 (95% CI ‐0.09 to 0.72, P = 0.13) (Table 1). (summary of findings Table for the main comparison).
| Outcome | Studies | Participants | Statistical method | Effect estimate |
| Change in participation (using the Rotterdam Handicap Scale) > 12 weeks after commencement of intervention | 1 (cross‐over) | 74 | Mean difference (IV, Fixed, 95% CI) | 0.31 [‐0.09 to 0.72] |
Fatigue after 12 or more weeks
Not reported at this time point.
Activity limitations after 12 or more weeks
Not reported at this time point.
Participation restrictions after 12 or more weeks
Not reported at this time point.
Health‐related quality of life (HRQoL) after 12 or more weeks
Not reported at this time point.
Adverse events
Reported in Garssen 2006.
No serious adverse events were reported. However, mild, transient adverse effects were reported in 32 out of 80 (40%) participants: 22 participants during amantadine treatment and 10 during placebo treatment. Adverse effects included anticholinergic complaints (six participants: three in each treatment group), gastrointestinal complaints (10 participants: seven in placebo group and three in amantadine group), sleep complaints (10 participants: nine in amantadine group and one in placebo group) and other less frequent side effects (headache, feelings of nervousness and vivid dreams). No side effects were reported to have affected trial participation.
Ascorbic acid versus placebo in Charcot‐Marie‐Tooth disease
Primary outcome
Fatigue between four and 12 weeks after commencement of intervention
Neither study evaluating ascorbic acid for Charcot‐Marie‐Tooth disease 1A reported change in fatigue within the time scale of the primary outcome for this review (Micallef 2009; Pareyson 2011).
Secondary outcomes
Data for secondary outcomes were reported at 12 months after commencement of the intervention in Micallef 2009 and at 24 months after commencement of the intervention in Pareyson 2011. Where two different doses of ascorbic acid were used in Micallef 2009, we combined data from both intervention arms, including participants receiving a higher dose (3 g ascorbic acid) and those receiving a lower dose (1 g ascorbic acid) in comparisons for all outcomes.
Fatigue after more than 12 weeks
Reported in Micallef 2009 and Pareyson 2011.
Both studies assessed fatigue using change in Visual Analogue Scale for fatigue (VAS‐F). Fatigue was assessed at 12‐month follow‐up using a 0 to 100 VAS for fatigue in Micallef 2009 and at 24‐month follow‐up using a 0 to 10 VAS for fatigue in Pareyson 2011. The pooled estimate of effect of the change in fatigue more than 12 weeks after commencement of the intervention was standardised mean difference (SMD) ‐0.12 (95% CI ‐0.32 to 0.08, P = 0.25; N = 404; I² = 0%) (see Analysis 1.1; Figure 2).
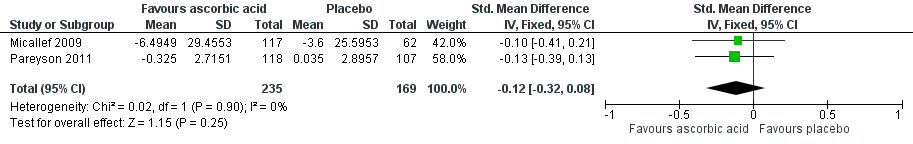
Forest plot of comparison: 2 Ascorbic acid versus placebo, outcome: 2.1 Change in fatigue > 12 weeks after commencement of intervention at 12 months [VAS 0 ‐ 100).
Activity limitations after 12 or more weeks
Reported in Micallef 2009 and Pareyson 2011.
Activity limitation was assessed using the ODSS at 12 months after commencement of intervention in Micallef 2009 and the Overall Neuropathy Limitations Scale (ONLS) 24 months after commencement of intervention in Pareyson 2011. Pooling of data in a meta‐analysis was not possible since data for ordinal scale scores were presented as medians plus interquartile ranges (IQRs), and there was no indication of whether the data were skewed. However, the authors reported that there was no significant difference (P = 0.22) in median change in activity limitation between placebo and 1 g or 3 g doses of ascorbic acid at 12 months in Micallef 2009, and no significant difference (P = 0.99) in median change in activity limitation between placebo and 1.5 g dose of ascorbic acid in Pareyson 2011.
In addition, further measures of activity limitation were included in both studies. The 10MWT was measured in both Micallef 2009 and Pareyson 2011 (at 12 months and 24 months, respectively) and the pooled estimate of effect for change in time taken to walk 10 metres more than 12 weeks after commencement of the intervention was MD ‐0.39 (95% CI ‐1.04 to 0.26, P = 0.24; N = 406; I² = 0%) (Analysis 1.2; Figure 3). In addition the 9HPT was measured in Pareyson 2011 at 24 months and showed that there was no significant effect of ascorbic acid (MD ‐0.74, 95% CI ‐1.60 to 0.12, P = 0.09; N = 224) (Analysis 1.3).
![Forest plot of comparison: 1 Ascorbic acid versus placebo, outcome: 1.2 Change in 10 m timed walk > 12 weeks after commencement of the intervention (measured at 12 to 24 months) [seconds].](/cdsr/doi/10.1002/14651858.CD008146.pub2/media/CDSR/CD008146/image_n/nCD008146-AFig-FIG03.png)
Forest plot of comparison: 1 Ascorbic acid versus placebo, outcome: 1.2 Change in 10 m timed walk > 12 weeks after commencement of the intervention (measured at 12 to 24 months) [seconds].
Participation restrictions after 12 or more weeks
Not reported in Micallef 2009 or Pareyson 2011.
Health‐related quality of life (HRQoL) after 12 or more weeks
Reported as an outcome and assessed using the SF‐36 at 12 months after commencement of the intervention in Micallef 2009 and 24 months after commencement of the intervention in Pareyson 2011. We calculated a pooled estimate of the change in physical functioning, as measured by the Physical Component Score (PCS) in Micallef 2009 and the Physical Function Scale (PFS) of the SF‐36 in Pareyson 2011. Using data from 400 participants, the SMD was 0.08 (95% CI ‐0.12 to 0.28) favouring placebo (see Figure 4, Analysis 1.4). There was no significant statistical heterogeneity, despite the difference in duration of follow‐up (I² = 0%, P = 0.67).
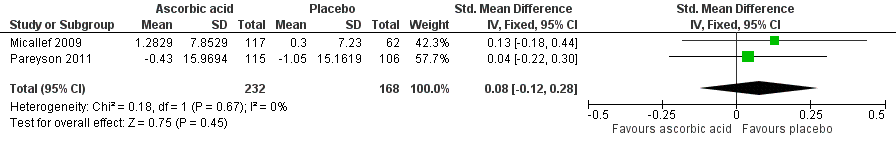
Forest plot of comparison: 2 Ascorbic acid versus placebo, outcome: 2.4 Change in physical function score of SF‐36.
Micallef 2009 reported HRQoL assessed by the Mental Component Score of the SF‐36 and for this too there was no significant difference (P = 0.77) between either dose of ascorbic acid versus placebo or in a combined analysis, between both doses of ascorbic acid versus placebo, 12 months after the start of treatment (Analysis 1.5).
Adverse events
Reported in Micallef 2009 and Pareyson 2011.
In Micallef 2009, 896 adverse events were documented in 158 (89%) participants (55 of 62 participants in the placebo group (89%); 47 of 56 participants in the 1 g ascorbic acid group (89%), and 56 of 61 participants in the 3 g ascorbic acid group (92%)), of which only 24 were serious adverse events. Serious adverse events were recorded in 22 participants (eight in the placebo group, seven in the 1 g ascorbic acid group and seven in the 3 g ascorbic acid group). Researchers deemed the majority of events (70%) not to be related to treatment and 208 (25%) to have a possible relation to treatment, of which only 12 (11 listed in the report) were severe: upper abdominal pain (2), dyspepsia (2), headache (1), insomnia (1), muscle spasm (1), musculoskeletal stiffness (1), rash (1), rhinitis (1) and sleep disturbance (1). Six participants (one in the placebo group, four in the 1 g ascorbic acid group and one in the 3 g ascorbic acid group) gave adverse events (cystitis, myalgia, depression, colitis and insomnia) as reasons for withdrawal from the study.
In Pareyson 2011, 21 serious adverse events were documented in 20 participants (eight events in seven of 138 participants in the ascorbic acid group and 13 events in 13 of 133 participants in the placebo group). Researchers considered all but one event, where a woman in the placebo group was admitted to hospital due to anaemia, as unrelated to the intervention. Thirteen participants experiencing adverse events were lost to follow‐up (six in the placebo group and seven in the ascorbic acid group), and eight participants (six in the placebo group and two in the ascorbic acid group) ceased treatment as a result of a serious adverse event. Five of these eight continued with follow‐up and three (two receiving placebo and one receiving ascorbic acid) withdrew from the study. Adverse events in those who discontinued the interventions included gastralgia, abdominal cramps or diarrhoea (9), surgery (4), renal colic (1), pregnancy (3), rectal bleeding (1), saccharine intolerance (1) and gum disease (1). The pooled risk ratio (RR) for serious adverse events in studies of ascorbic acid for CMT1A was 0.70 (95% CI 0.39 to 1.27, P = 0.24; N = 450; I² = 0%) (see Analysis 1.6, Figure 5), indicating no significant difference in serious adverse events between ascorbic acid and placebo.
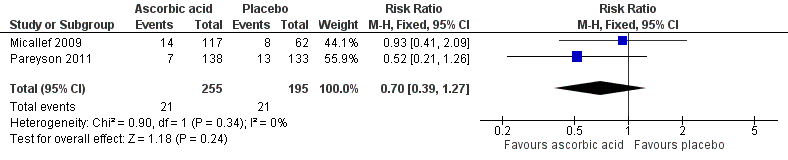
Forest plot of comparison: 2 Ascorbic acid versus placebo, outcome: 2.6 Adverse events.
Discussion
Summary of main results
Our search identified three randomised controlled trials (RCTs) that met the inclusion criteria. The trials evaluated the effect of two different drugs on fatigue in two different populations of people with peripheral neuropathy. Two studies examined the effect of different doses of ascorbic acid in Charcot‐Marie‐Tooth disease 1A, and therefore no combined analysis was possible.
The first study (Garssen 2006) was a block‐randomised, double‐blind, placebo‐controlled, cross‐over study of amantadine for severe fatigue in 74 neurologically stable participants at least six months after Guillain‐Barré syndrome. We judged the risk of bias of the trial to be low. The study reported a moderate reduction in fatigue severity scale (FSS) score for participants treated with amantadine but the confidence interval was wide and study size relatively small, suggesting that the study may have been underpowered to detect a difference; we therefore graded the quality of the evidence as low. These data indicate that larger studies might improve the power to detect a reduction in risk of fatigue with amantadine. Nevertheless, the mean difference (MD) did not exceed the minimal clinically important difference for change in FSS scores estimated for people with systemic lupus erythematosus (MD 0.6; 95% CI 0.3 to 0.9), indicating that it is likely that there is no effect of amantadine compared with placebo in the treatment of severe fatigue (see summary of findings Table for the main comparison). A significant decrease in fatigue was noted between two pre‐treatment baseline visits two weeks apart. Since the participants were considered to be neurologically stable, this suggests that increased attention to people with fatigue due to Guillain‐Barré syndrome may improve symptoms of fatigue.
The two studies evaluating ascorbic acid for Charcot‐Marie‐Tooth (CMT) disease 1A were block‐randomised, double‐blind, placebo‐controlled trials of different doses of ascorbic acid for the treatment of Charcot‐Marie‐Tooth disease 1A (Micallef 2009; Pareyson 2011). The risk of bias for Micallef 2009 was low, despite the fact that there was no report of how missing data were dealt with in the intention‐to‐treat analysis. All other aspects of risk of bias assessment were adequate. The main findings were that whilst ascorbic acid was well‐tolerated and safe for adults with Charcot‐Marie‐Tooth disease 1A over a period of 12 months, there were no significant differences in effect across all outcomes, including fatigue, between placebo, 1 g or 3 g of ascorbic acid (see summary of findings Table 2). There was a low risk of bias in the remaining study (Pareyson 2011), and the main findings were that ascorbic acid supplementation at a dose of 1.5 g per day for 24 months was well tolerated but there was no significant effect on neuropathy or secondary outcomes compared with placebo.
Overall completeness and applicability of evidence
Only three RCTs (Garssen 2006; Micallef 2009; Pareyson 2011) were eligible for inclusion in the review. One was of amantadine for severe fatigue in Guillain‐Barré syndrome and the others were of three different doses of ascorbic acid for people with Charcot‐Marie‐Tooth disease 1A. RCTs evaluating the effect of other interventions for fatigue, such as other drugs; pacing and grading of physical activity; general or specific exercise; compensatory strategies such as orthotics; relaxation; counselling; cognitive and educational strategies; and interventions for fatigue that form part of a multidimensional programme, or trials of interventions for other causes of peripheral neuropathy, were not available. None of the included studies considered the cost effectiveness of interventions for fatigue in peripheral neuropathy
Quality of the evidence
The overall risk of bias in the included trials (Garssen 2006; Micallef 2009; Pareyson 2011) was low.
Garssen 2006 reported a double‐blind, placebo‐controlled study, where, despite a block randomisation procedure affecting bias in sequence generation and incomplete reporting of secondary outcomes, there was effective allocation concealment and P values for secondary outcomes were available. Reporting of data for fatigue during both phases of the trial indicates that there was no significant carry‐over effect. We therefore judged the overall risk of bias to be low.
Micallef 2009 reported a block‐randomised, double‐blind, placebo‐controlled study, where all assessed aspects of risk of bias were adequate except for the lack of reporting of how missing data were dealt with for an intention‐to‐treat analysis. We therefore judged the overall risk of bias to be low.
Pareyson 2011 reported a block‐randomised, double‐blind, placebo‐controlled study with a low risk of bias overall.
Since only one trial of amantadine for the treatment of fatigue in Guillain‐Barré syndrome (Garssen 2006) and two trials of ascorbic acid for treatment of Charcot‐Marie‐Tooth disease 1A (Micallef 2009; Pareyson 2011) are included in the review, it is not possible to address the review objectives for drug or other interventions. The Garssen 2006 study was relatively small (74 participants) and the results actually suggest that there may be an effect, but the study is under‐powered. We downgraded the evidence from this study to low because of its imprecision (summary of findings Table for the main comparison). Larger studies including those with different doses and frequency of administration of amantadine are desirable before a definite conclusion about the effect of amantadine to treat fatigue in peripheral neuropathy and in particular in Guillain‐Barré syndrome can be given.
The studies of ascorbic acid for Charcot‐Marie‐Tooth disease 1A were larger, and evaluated the effect of three different doses of ascorbic acid in Charcot‐Marie‐Tooth disease 1A (Micallef 2009; Pareyson 2011). The lack of reporting of how missing data were dealt with in Micallef 2009 means that it is difficult to assess whether the risk of bias is likely to have favoured the control or ascorbic acid treatment groups in this study . Nevertheless, the low risk of bias associated with the final study (Pareyson 2011) and the fact that it has similar findings to Micallef 2009 meant that we did not downgrade the evidence because of trial design or conduct, but did downgrade it because of the lack of reporting of how missing data was dealt with in Micallef 2009. This means that there is moderate‐quality evidence suggesting that ascorbic acid has no benefit in the treatment of fatigue in CMT (See summary of findings Table 2).
The primary outcome of fatigue was evaluated using the FSS or VAS for fatigue in the included studies. The FSS is based on classical test theory (CTT), where scales may include items not relevant for particular clinical populations, and scale sum scores assume equal weighting of items where this has not necessarily been demonstrated. This potentially introduces bias into the assessment of the underlying outcome of interest. However, evidence in the current review was not downgraded due to directness based on outcomes used, since the effect of indirectness is not currently known. Nevertheless, clinicians and researchers are moving towards linearly weighted scales based on modern scientific methods such as Rasch models or item response theory (IRT) for evaluation of outcomes in clinical populations. The FSS has recently been fully evaluated and modified using the Rasch unidimensional measurement model (RUMM2020) and is recommended for use in immune‐mediated neuropathy (van Nes 2009). Its relevance for other peripheral neuropathies remains to be evaluated.
Potential biases in the review process
Two review authors were also investigators in an included trial (Garssen 2006), but neither was involved in assessing the risk of bias in this study.
We contacted study authors where there were missing data, and these was provided by Garssen 2006 for one secondary outcome. We also contacted authors of published abstracts with limited reporting of data for further information, but they either did not reply or declined to, stating their intention to publish a full report of the study in the future as a reason.
This systematic review used an adequate search strategy to avoid missing any RCTs on treatments for fatigue in peripheral neuropathy. There were no language restrictions in our searches, and whilst a limitation of the review is that we did not initially plan in our protocol to search clinical trials registers, this additional search was subsequently conducted; this difference between the protocol and review is flagged up below.
The criteria for types of participants for including studies accepted the diagnosis of peripheral neuropathy offered by the authors, provided that it stipulated the presence of clinical impairment characteristic of peripheral neuropathy. We considered only sensory, motor or combined sensory and motor peripheral neuropathies, and excluded studies of participants with focal entrapment neuropathies with pain as the primary presenting feature, and with poliomyelitis. There is therefore a small possibility that misdiagnosis of participants may have affected study inclusion, and that studies with mixed populations of participants that include both sensory, motor and combined sensory motor neuropathies as well as painful entrapment neuropathies or poliomyelitis were excluded. This could be a potential source of selection bias in the review.
Agreements and disagreements with other studies or reviews
There are currently no other systematic reviews of interventions for fatigue in peripheral neuropathy available, despite many treatments for fatigue including: drugs, pacing and grading of physical activity, general or specific exercise, compensatory strategies such as orthotics, relaxation, counselling, cognitive and educational strategies, either alone or as part of a multidimensional programme.
Systematic reviews of interventions for fatigue in other conditions
In the absence of evidence from reviews or high‐quality RCTs in peripheral neuropathy it may be helpful to consider the effect of interventions for fatigue in other conditions, including chronic fatigue syndrome (CFS), fibromyalgia, multiple sclerosis and cancer‐related fatigue. The high incidence of anxiety and depression in functional somatic syndromes such as fibromyalgia and CFS (Henningsen 2003) and the specific pathophysiological features of multiple sclerosis and cancer‐related fatigue mean that recommendations from systematic reviews should be considered with caution. In all cases we included only evidence from Cochrane systematic reviews.
Drugs for fatigue
The evidence from Cochrane systematic reviews of drugs in the treatment of fatigue in other conditions was of varying quality. A systematic review of serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia indicated that they only provide a small improvement in fatigue (1.7% improvement) over placebo (Hauser 2013). In reviews of amantadine or carnitine for fatigue in multiple sclerosis, the poor quality of the available RCTs meant that no conclusion regarding their efficacy could be drawn (Pucci 2009; Tejani 2012). Finally, whilst the updated systematic review of drug therapy for the management of cancer‐related fatigue provides evidence that psychostimulant trials may give a clinically meaningful improvement in cancer‐related fatigue, new safety data indicate that the drugs are associated with increased adverse outcomes and they can no longer be recommended in the treatment of cancer‐related fatigue (Minton 2010).
Non‐drug interventions for fatigue
We reviewed the evidence from recent Cochrane systematic reviews of non‐drug interventions for fatigue in other conditions, which suggests that exercise (Edmonds 2004) and cognitive behavioural therapy (CBT) (Price 2008) for fatigue in CFS are both better than usual care, with the evidence for CBT being more extensive and robust than for exercise. In a review of exercise for treating fibromyalgia syndrome, there is very limited evidence that aerobic‐only exercise is likely to have no effect on fatigue in individuals with fibromyalgia (Busch 2007). A systematic review of cognitive behavioural therapies for fibromyalgia concludes that there was only a 5.8% (95% CI 0.05 to 11.3) effect on fatigue (Bernardy 2013). A systematic review of exercise for management of cancer‐related fatigue concluded that aerobic exercise is beneficial (Cramp 2012).
Evidence from individual studies of interventions for fatigue in neuropathy
One article describing an uncontrolled case series of people with Charcot‐Marie‐Tooth disease 1A treated with modafinil for fatigue reported that all four participants had less fatigue within at least 10 days of commencing modafinil treatment (Carter 2006). The case study design means that this report has substantial risk of bias but may suggest that modafinil is a potentially useful drug for fatigue in people with Charcot‐Marie‐Tooth disease 1A, and merits well‐designed trials.
The wide variety of causes and consequences in peripheral neuropathy, combined with a relatively low prevalence of individual conditions means that controlled trials of non‐drug interventions have been limited. In addition, despite the substantial prevalence of fatigue in people with peripheral neuropathy (Merkies 1999), subjective fatigue is not routinely evaluated in RCTs and other study designs of such interventions. However, one cohort study (Garssen 2004) and one case‐control study (Graham 2007) of exercise for people with Guillain‐Barré syndrome and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) reported significant reductions in fatigue after 12 weeks of either supervised aerobic cycling (Garssen 2004) or unsupervised community‐based exercise (Graham 2007). Whilst these studies are subject to very high risks of bias due to their design, they may suggest that RCTs of exercise for people with fatigue due to peripheral neuropathy should be conducted.
Methodological quality summary: review authors' judgements about each methodological quality item for each included study. Key: green (+) = low risk of bias; yellow (?) = unclear risk of bias; red (‐) = high risk of bias.
Forest plot of comparison: 2 Ascorbic acid versus placebo, outcome: 2.1 Change in fatigue > 12 weeks after commencement of intervention at 12 months [VAS 0 ‐ 100).
Forest plot of comparison: 1 Ascorbic acid versus placebo, outcome: 1.2 Change in 10 m timed walk > 12 weeks after commencement of the intervention (measured at 12 to 24 months) [seconds].
Forest plot of comparison: 2 Ascorbic acid versus placebo, outcome: 2.4 Change in physical function score of SF‐36.
Forest plot of comparison: 2 Ascorbic acid versus placebo, outcome: 2.6 Adverse events.
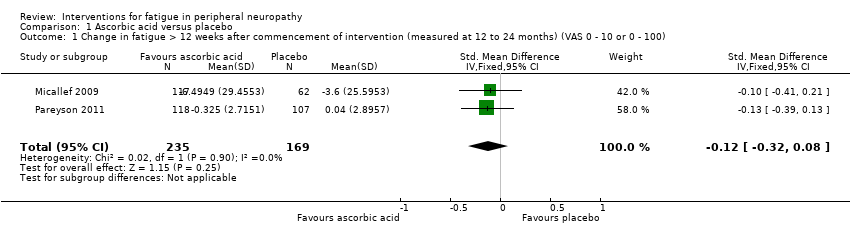
Comparison 1 Ascorbic acid versus placebo, Outcome 1 Change in fatigue > 12 weeks after commencement of intervention (measured at 12 to 24 months) (VAS 0 ‐ 10 or 0 ‐ 100).

Comparison 1 Ascorbic acid versus placebo, Outcome 2 Change in 10 m timed walk > 12 weeks after commencement of the intervention (measured at 12 to 24 months).

Comparison 1 Ascorbic acid versus placebo, Outcome 3 Change in speed of completion of 9 hole peg test > 12 weeks after commencement of intervention (measured at 24 months).

Comparison 1 Ascorbic acid versus placebo, Outcome 4 Change in physical function score of SF‐36 > 12 weeks after commencement of the intervention (measured at 12 to 24 months).

Comparison 1 Ascorbic acid versus placebo, Outcome 5 Change in mental function score of SF‐36 > 12 weeks after commencement of the intervention (measured at 12 months).
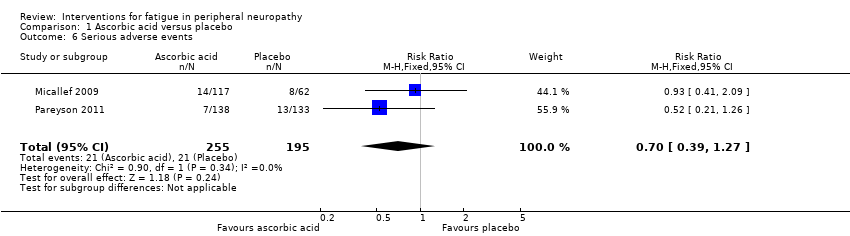
Comparison 1 Ascorbic acid versus placebo, Outcome 6 Serious adverse events.
| Amantadine versus placebo for fatigue in Guillain‐Barré syndrome | ||||||
| Patient or population: people with fatigue in Guillain‐Barré syndrome | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Amantadine | |||||
| Reduction in fatigue of at least 1‐point on the Fatigue Severity Scale between 4 to 12 weeks after commencement of intervention | 257 per 1000 | 162 per 1000 | OR 0.56 | 74 | ⊕⊕⊝⊝ | ‐ |
| Change in fatigue 12 or more weeks after commencement of intervention (not measured) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in activity limitation 12 or more weeks after commencement of intervention (not measured) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in participation 12 or more weeks after commencement of intervention using the | The mean change in participation 12 or more weeks after commencement of intervention using the control group was | The mean change in participation 12 or more weeks after commencement of intervention using the intervention group was | 74 | ⊕⊕⊝⊝ | ‐ | |
| Health related quality of life after 12 or more weeks or more after commencement of intervention (not reported) | ‐ | ‐ | ‐ | ‐ | ‐ | EHQ and SF‐36 measured but data not fully reported |
| Serious adverse events | See comment | See comment | Not estimable | 80 | See comment | 0 participants had serious adverse events in this study |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Evidence downgraded two levels due to imprecision (small sample size and wide CI that crosses the line of no effect). | ||||||
| Ascorbic acid versus placebo for fatigue in peripheral neuropathy | ||||||
| Patient or population: people with fatigue in peripheral neuropathy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Ascorbic acid | |||||
| Reduction in fatigue of at least 1 point on the Fatigue Severity Scale (FSS) between 4 to 12 weeks after commencement of intervention (not measured) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in fatigue 12 or more weeks after commencement of intervention | ‐ | The mean change in fatigue 12 or more weeks after commencement of intervention in the intervention groups was | ‐ | 404 | ⊕⊕⊕⊝ | ‐ |
| Change in activity limitation (ODSS/ONLS) 12 or more weeks after commencement of intervention | See comment | See comment | Not estimable | 406 | ‐ | Data available were not suitable for meta‐analysis |
| Change in 10 metre timed walk 12 or more weeks after commencement of intervention | The mean changes in 10 metre timed walk 12 or more weeks after commencement of intervention in the control groups werea decrease of 0.40 seconds and an increase of 0.76 seconds2 | The mean change in 10 metre timed walk 12 or more weeks after commencement of intervention in the intervention groups was a decrease of | 406 | ⊕⊕⊕⊝ | ‐ | |
| Change in speed of completion of 9‐hole peg test (in seconds) 12 or more weeks after commencement of intervention | The mean change in speed of completion of the 9‐hole peg test was an increase of 0.85 seconds in the control group | The mean change in speed of completion of the 9‐hole peg test was a decrease of 0.74 seconds (1.60 faster to 0.12 slower) | Not estimable | 224 | ⊕⊕⊕⊝ | ‐ |
| Health‐related quality of life 12 or more weeks after commencement of the intervention Change in SF‐36 physical function score | ‐ | The mean health‐related quality of life 12 or more weeks after commencement of the intervention (change in SF‐36 physical function score) in the intervention groups was | ‐ | 400 | ⊕⊕⊕⊝ | ‐ |
| Change in participation 12 or more weeks after commencement of intervention (not measured) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Health‐related quality of life 12 or more weeks after commencement of the intervention Change in mental function score of SF‐36 | The mean change in health‐related quality of life 12 or more weeks after commencement of the intervention (change in mental function score of SF‐36) in the control group was 1.2 points | The mean health‐related quality of life 12 or more weeks after commencement of the intervention (change in mental function score of SF‐36) in the intervention groups was | ‐ | 123 | ⊕⊕⊕⊝ | ‐ |
| Serious adverse events | 108 per 1000 | 76 per 1000 | RR 0.70 | 450 | ⊕⊕⊕⊕ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Only one or two studies included. | ||||||
| Outcome | Studies | Participants | Statistical method | Effect estimate |
| Change in participation (using the Rotterdam Handicap Scale) > 12 weeks after commencement of intervention | 1 (cross‐over) | 74 | Mean difference (IV, Fixed, 95% CI) | 0.31 [‐0.09 to 0.72] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in fatigue > 12 weeks after commencement of intervention (measured at 12 to 24 months) (VAS 0 ‐ 10 or 0 ‐ 100) Show forest plot | 2 | 404 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.12 [‐0.32, 0.08] |
| 2 Change in 10 m timed walk > 12 weeks after commencement of the intervention (measured at 12 to 24 months) Show forest plot | 2 | 406 | Mean Difference (IV, Fixed, 95% CI) | ‐0.39 [‐1.04, 0.26] |
| 3 Change in speed of completion of 9 hole peg test > 12 weeks after commencement of intervention (measured at 24 months) Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4 Change in physical function score of SF‐36 > 12 weeks after commencement of the intervention (measured at 12 to 24 months) Show forest plot | 2 | 400 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.08 [‐0.12, 0.28] |
| 5 Change in mental function score of SF‐36 > 12 weeks after commencement of the intervention (measured at 12 months) Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6 Serious adverse events Show forest plot | 2 | 450 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.39, 1.27] |