Preparados de valproato para la agitación en la demencia
Resumen
Antecedentes
Se ha informado de agitación en hasta el 90% de los pacientes con demencia. La agitación en los pacientes con demencia empeora la carga de los cuidadores, aumenta el riesgo de lesiones y aumenta la necesidad de internamiento. Los preparados de valproato se han utilizado para intentar controlar la agitación en la demencia, pero se ha cuestionado su seguridad y eficacia.
Objetivos
Determinar la eficacia y los efectos adversos de los preparados de valproato utilizados para tratar la agitación en los pacientes con demencia, incluido el impacto sobre los cuidadores.
Métodos de búsqueda
Se realizaron búsquedas en ALOIS, el Registro Especializado del Grupo Cochrane de Demencia y Trastornos Cognitivos (Cochrane Dementia and Cognitive Improvement Group), el 7 de diciembre 2017, mediante los términos: valproic OR valproate OR divalproex. ALOIS contiene registros de todas las bases de datos principales de atención sanitaria (The Cochrane Library, MEDLINE, EMBASE, PsycINFO, CINAHL, LILACS), así como de muchas bases de datos de ensayos y fuentes de literatura gris.
Criterios de selección
Ensayos aleatorizados controlados con placebo que evaluaron las preparaciones de valproato para la agitación en pacientes con demencia.
Obtención y análisis de los datos
Dos autores de la revisión de forma independiente examinaron los estudios recuperados en función de los criterios de inclusión y extrajeron los datos y evaluaron la calidad metodológica de los estudios incluidos. Cuando fue necesario, se estableció contacto con los autores de los ensayos para obtener datos adicionales, incluidas las subescalas relevantes o para obtener otra información faltante. Los datos se agruparon en metanálisis cuando fue posible. Ésta es una actualización de una revisión Cochrane publicada en 2009. No se encontraron estudios nuevos para su inclusión.
Resultados principales
La revisión incluyó cinco estudios con 430 participantes. Los estudios variaron en cuanto a los preparados de valproato, las dosis medias (480 mg/día a 1000 mg/día), la duración del tratamiento (de tres a seis semanas) y las medidas de resultados utilizadas. En general, los estudios se realizaron de manera adecuada, aunque faltó alguna información metodológica y uno de ellos presentó alto riesgo de sesgo de desgaste.
La calidad de la evidencia relacionada con el resultado de eficacia primaria de agitación varió de moderada a muy baja. Se encontró evidencia de calidad moderada en dos estudios que midieron el comportamiento con la Brief Psychiatric Rating Scale (BPRS) (rango 0 a 108) y con el factor de agitación de la BPRS (rango 0 a 18). Se encontró que probablemente hubo poco o ningún efecto del tratamiento con valproato a las seis semanas (BPRS total: diferencia de medias [DM] 0,23; intervalo de confianza [IC] del 95%: ‐2,14 a 2,59; 202 participantes, dos estudios; factor de agitación de la BPRS: DM ‐0,67; IC del 95%: ‐1,49 a 0,15; 202 participantes; dos estudios). La evidencia de calidad muy baja de tres estudios que midieron la agitación con el Cohen‐Mansfield Agitation Index (CMAI) fue consistente con la falta de efecto del tratamiento con valproato sobre la agitación. Hubo evidencia de calidad variable de otros resultados de comportamiento informados en estudios individuales, de que no había diferencias entre los grupos o que hubo un efecto beneficioso para el grupo placebo.
Tres estudios que midieron la función cognitiva utilizando el Mini‐Mental State Examination (MMSE), encontraron poco o ningún efecto del valproato a las seis semanas, pero no hay certeza acerca de este resultado porque la calidad de la evidencia fue muy baja. Dos estudios que evaluaron la capacidad funcional mediante la Physical Self‐Maintenance Scale (PSMS) (rango 6 a 30) encontraron que probablemente hubo una función ligeramente peor en el grupo tratado con valproato, que fue de importancia clínica poco clara (DM 1,19; IC del 95%: 0,40 a 1,98; 203 participantes, dos estudios; evidencia de calidad moderada).
El análisis de los efectos adversos y los eventos adversos graves (EAG) indicó una mayor incidencia en los participantes tratados con valproato. Un metanálisis de tres estudios mostró que puede haber habido una mayor tasa de efectos adversos entre los participantes tratados con valproato que entre los controles (odds ratio [OR] 2,02; IC del 95%: 1,30 a 3,14; 381 participantes, tres estudios, evidencia de calidad baja). El análisis agrupado del número de EAG de los dos estudios que informaron estos datos indicó que los participantes tratados con preparados de valproato tuvieron más probabilidades de experimentar EAG (OR 4,77; IC del 95%: 1,00 a 22,74; 228 participantes, dos estudios), pero la calidad muy baja de los datos dificultó establecer cualquier conclusión firme con respecto a los EAG. Los eventos adversos individuales que fueron más frecuentes en el grupo tratado con valproato incluyeron sedación, síntomas gastrointestinales (náuseas, vómitos y diarrea) e infecciones urinarias.
Conclusiones de los autores
Esta revisión actualizada corrobora los hallazgos anteriores de que las preparaciones de valproato probablemente no son efectivas para tratar la agitación en los pacientes con demencia, y que se asocian con una mayor tasa de efectos adversos y posiblemente de EAG. Sobre la base de esta evidencia, el tratamiento con valproato no se puede recomendar para el tratamiento de la agitación en la demencia. Tal vez no se justifique la realización de nuevos estudios de investigación, en particular a la luz del mayor riesgo de que se produzcan efectos adversos en este grupo de pacientes, a menudo frágiles. Los estudios de investigación se deberían centrar en cambio en intervenciones no farmacológicas efectivas para este grupo de pacientes o, en situaciones en las que se pueda necesitar medicación, y en estudios adicionales sobre cómo utilizar otros medicamentos de la forma más efectiva y segura posible.
PICO
Resumen en términos sencillos
Preparados de valproato para el tratamiento del comportamiento agitado en pacientes con demencia
Antecedentes
El comportamiento agitado es muy frecuente en las últimas etapas de la demencia. Puede incluir conductas verbales como gritos, y conductas físicas como deambular o agresión física. Se ha demostrado que empeora el estrés que experimentan los cuidadores familiares, aumenta el riesgo de lesiones y aumenta la necesidad de que los pacientes con demencia se trasladen a la atención institucional.
Un tipo de medicamento que se ha utilizado para tratar el comportamiento agitado de los pacientes que padecen demencia es el valproato, que está disponible en varios preparados diferentes (ácido valproico, divalproex sódico, valproato sódico y valproato semisódico). Estos medicamentos no se recomiendan en las guías actuales (p.ej. las del National Institute for Health and Care Excellence), pero en ocasiones se les administran a los pacientes con demencia para tratar el comportamiento agitado.
Propósito de esta revisión
Se deseaba examinar la evidencia sobre la efectividad y la seguridad de la administración de preparados de valproato a pacientes con demencia para tratar la agitación.
Estudios incluidos en esta revisión
Se buscó en las bases de datos médicas hasta diciembre 2017 los estudios que compararon cualquier preparación de valproato con placebo (tableta simulada) para tratar el comportamiento agitado en pacientes a los que se les diagnosticó demencia.
Se incluyeron cinco estudios con 479 participantes que presentaban diversos tipos de demencia y comportamiento agitado. La mayoría de los estudios duraron seis semanas, aunque uno solo duró tres semanas. En general, los estudios se realizaron de manera adecuada, pero no siempre los métodos se informaron de manera completa y un estudio tuvo alto riesgo de sesgo debido al gran número de pacientes que abandonaron el grupo tratado con valproato.
Hallazgos clave
Los estudios midieron el comportamiento agitado con varias escalas y la fiabilidad de la evidencia de las diferentes escalas varió de moderada a muy baja. En general, no se encontró evidencia de que los preparados de valproato mejoraran el comportamiento, o específicamente, el comportamiento agitado. Se encontró que las preparaciones de valproato probablemente tuvieron poco o ningún efecto sobre la capacidad de los participantes para realizar las actividades diarias. No fue posible asegurar que tuvieran un efecto sobre la cognición (el pensamiento y el recuerdo) porque la fiabilidad de la evidencia fue muy baja.
Se encontró evidencia de baja fiabilidad en tres estudios de que los participantes que toman valproato pueden tener más probabilidades de experimentar efectos perjudiciales que los que toman placebo. No fue posible asegurar que hubiera diferencias en cuanto a los daños graves como las enfermedades graves o el ingreso hospitalario, pero los datos de dos estudios indicaron que éstos eventos podrían ser más frecuentes en los participantes que tomaron valproato. Algunos de los efectos secundarios asociados con el valproato fueron la somnolencia, la sensación de malestar, las náuseas, las deposiciones acuosas y las infecciones urinarias.
Conclusiones
Solo se identificaron cinco estudios relativamente pequeños para su inclusión en esta revisión. Variaron en cuanto a sus métodos, el tipo de medicamento y su dosis, la duración del tratamiento y las escalas utilizadas para hacer las mediciones. Lo anterior limitó la capacidad para agrupar los datos de todos los estudios. Sin embargo, se podría confiar moderadamente en la conclusión de que los preparados de valproato no mejoran el comportamiento agitado en la demencia. También se pueden asociar con efectos nocivos.
Authors' conclusions
Summary of findings
| Valproate preparations compared to placebo for agitation in dementia | ||||||
| Patient or population: people with agitation in dementia | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with valproate preparations | |||||
| Agitation and aggression Scale: 0–108 (higher score indicated higher level of dysfunction) | The mean change from baseline for agitation and aggression was –5.34 points. | MD 0.23 higher | — | 202 | ⊕⊕⊕⊝ | — |
| Agitation and aggression Scale: 0–18 (higher score indicated higher level of dysfunction) | The mean change from baseline for agitation and aggression was –1.88 points. | MD 0.67 lower | — | 202 | ⊕⊕⊕⊝ | — |
| Agitation and aggression Scale: 0–216 (higher score indicated more agitated behaviour) | The mean change from baseline for agitation and aggression was –4.42 points. | MD 1.84 lower | — | 217 | ⊕⊝⊝⊝ | — |
| Cognition Scale: 0–30 (lower score indicated greater cognitive impairment) | The mean change from baseline for cognition was 0.46 points. | MD 0.7 lower | — | 217 | ⊕⊝⊝⊝ | — |
| Functional performance Scale: 6–30 (higher score indicated greater impairment in ADL) | The mean change from baseline for functional performance was 0.06 points. | MD 1.19 higher | — | 203 | ⊕⊕⊕⊝ | — |
| Any adverse event by 6 weeks | Study population | OR 2.02 | 381 | ⊕⊕⊝⊝ | — | |
| 602 per 1000 | 753 per 1000 | |||||
| Serious adverse events by 6 weeks | Study population | OR 4.77 | 228 | ⊕⊝⊝⊝ | — | |
| 18 per 1000 | 79 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ADL: activities of daily living; CI: confidence interval; ITT: intention to treat; MD: mean difference; OR: odds ratio; RCT: randomised controlled trial. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level for imprecision due to small number of participants. bDowngraded in light of imprecision due to confidence intervals including the potential for harm or benefit. cDowngraded one level due to inconsistency (heterogeneity between studies). dDowngraded one level due to study limitations (risk of bias). | ||||||
Background
Description of the condition
Agitation is reported in up to 90% of people with dementia (Alzheimer's Society 2011a). A widely accepted definition of agitation is: "inappropriate verbal, vocal, or motor activity that is not explained by needs or confusion per se" (Billig 1991; Cohen‐Mansfield 1989). The descriptors of agitation include wandering, crying out, aggressiveness, repetitive movements, and unco‐operative behaviour. Agitation in people with dementia worsens carer burden, increases the risk of injury, and adds to the need for institutionalisations (Livingstone 2014).
Description of the intervention
Current guidelines recommend that people with dementia who develop non‐cognitive symptoms or behaviours that cause them distress or challenge those who provide their care should first have a comprehensive assessment to determine likely causative factors, such as physical illness, depression, pain, adverse effects of medication, personal or psychosocial factors, or aspects of their physical environment. Appropriate steps should then be taken to address those factors, and a period of 'watchful waiting' should be observed, if possible, as in many cases symptoms will improve or resolve over four to six weeks (Alzheimer's Society 2011a). The guidelines also suggest that consideration should be given to providing individualised interventions such as aromatherapy or multisensory stimulation as there is some evidence of their clinical effectiveness (Livingstone 2014; NICE 2006). In fact, research has shown that just 10 minutes of one‐to‐one time each day can reduce behavioural and psychological symptoms associated with dementia (BPSD) (Alzheimer's Society 2011b).
The National Institute for Health and Care Excellence (NICE) guideline on supporting people with dementia and their carers suggests that people with dementia who present with non‐cognitive symptoms or challenging behaviour should be offered pharmacological intervention in the first instance "only if they are severely distressed or there is an immediate risk of harm to the person or others" and that a thorough assessment of possible causes of the behaviour should be carried out as soon as possible (NICE 2016). Drug treatment for the control of violence, aggression, and extreme agitation should be implemented with the aim of avoiding sedation and the use of high doses or combinations of drugs, and with careful monitoring of the person's physical condition and any adverse effects (NICE 2006).
If drug treatment of agitation is considered necessary, then the drug classes recommended by NICE, in order, are antipsychotics, acetylcholinesterase inhibitors, and memantine. There is some evidence of modest benefits of antipsychotics in around 50% of people with dementia, but they are associated with adverse effects such as sedation, parkinsonism, gait disturbance, dehydration, falls, chest infection, accelerated cognitive decline, and stroke, and they are associated with increased mortality in the long term (Alzheimer's Society 2011b; Maher 2011). The increased risk of cerebrovascular adverse events and death in this patient group resulted in a Medicines and Healthcare products Regulatory Agency (MHRA) warning that no antipsychotic should be used for this indication in dementia (except risperidone in some circumstances) (MHRA 2012). Risperidone is the only antipsychotic licensed for people with dementia, and guidelines recommend treatment should be used for no longer than 12 weeks. The evidence of benefit of other types of antipsychotics is more limited, and use for BPSD is off‐label. Acetylcholinesterase inhibitors and memantine are licensed for the treatment of cognitive symptoms in Alzheimer's disease and there is some evidence that these medications may positively impact on agitated behaviour, although there is no evidence that they specifically improve agitation (NICE 2006).
Other medications that have been used to treat agitated behaviour in people with dementia include benzodiazepines, hypnotics, antidepressants, and anticonvulsants. There is no evidence of benefit of benzodiazepines for this indication, and they carry increased risk of adverse effects (Bierman 2007). There is relatively little evidence relating to antidepressants for agitated behaviour in dementia; findings on efficacy are mixed and there is evidence of adverse effects (Porteinsson 2014; Seitz 2011). Among anticonvulsants, carbamazepine and valproate preparations have both been used widely.
How the intervention might work
Various valproate preparations are available: valproic acid, divalproex, sodium valproate, and valproate semi‐sodium. Suggested mechanisms by which valproic acid may have an impact on agitation include enhancement of the intracerebral neurotransmitting agent, gamma‐butyric acid (GABA), antimanic action, and mood stabilising effect (Lon 1995). Since 1996, a more readily tolerated compound of valproate, divalproex, has been used. This drug differs slightly from valproic acid in that peak blood flow levels occur later (three to six hours, compared with three hours), but the dosage and half‐life of this drug are identical to those of valproic acid. Sodium valproate is licensed for the treatment of epilepsy in standard‐release oral preparations, and in modified‐released preparations for various indications according to the preparation. Sodium valproate or valproate semi‐sodium is licensed for the treatment of manic episodes in bipolar disorder. None of the valproate preparations are licensed for the management of agitated behaviour in people with dementia; therefore, use of for this purpose is off‐label.
Adverse effects associated with valproate preparations include falls, gait disturbances, sedation, tremor, muscular weakness, depressed mood, gastrointestinal disorders (nausea, vomiting, constipation, and diarrhoea), urinary tract infections (UTI), and thrombocytopenia. The current NICE advice on the use of valproate preparations for the management of aggression, agitation, and behavioural disturbances in dementia states that current evidence suggests that such medications are no more effective than placebo, and that adverse effects are also more common in people taking them (NICE 2015).
Why it is important to do this review
This is an update of a Cochrane Review first published in 2004, and previously updated in 2009.
One summary of evidence published by NICE suggests that valproate preparations are no more effective than placebo for agitation in dementia (NICE 2015). Despite this guidance, valproate preparations are still sometimes being used in this patient group, perhaps because other drug options are not always effective and may be associated with adverse effects. This update is intended to apply current Cochrane methods to synthesise the evidence concerning use of valproate for agitation in dementia, and to assess the quality of this evidence, in order to inform decision‐making by carers, clinicians, researchers, and policy‐makers.
Objectives
To determine the efficacy and adverse effects of valproate preparations used to treat agitation in people with dementia, including the impact on carers.
Methods
Criteria for considering studies for this review
Types of studies
We included only randomised, placebo‐controlled trials. We excluded interrupted time series trials. Where studies used a cross‐over design, we included only data from the first part of the study.
Types of participants
We included participants of either sex and of any age, both inpatients and outpatients (with or without carers). Dementia should have been diagnosed according to the classifications provided by Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM‐IV) (APA 1994), International Classification of Diseases, 10th edition (ICD‐10) (WHO 1991), Diagnostic and Statistical Manual of Mental Disorders, 3rd edition (DSM‐III) (APA 1980), or Diagnostic and Statistical Manual of Mental Disorders, 3rd revised edition (DSM‐IIIR) (APA 1987). In the absence of these criteria, we also accepted other evidence of dementia such as the Mini‐Mental State Examination (MMSE) (Folstein 1975), psychiatric evaluation, psychological evaluation, or a medical evaluation. We accepted definitions of agitation provided by individual investigators.
Because agitation is common in delirium, we had initially specified that all studies should have included clinical evaluation to rule out delirium and other treatable causes of agitation (e.g. pain, infection, drug effect, urinary or faecal retention) prior to entering people into the trial. However, reporting of baseline clinical evaluation was not always specific or detailed. Therefore, we took a pragmatic approach to avoid risking the loss of relevant evidence and included studies despite this information not being explicitly reported.
Types of interventions
We required at least one week of treatment with valproate preparations, of any dosage given by mouth, compared with placebo. People receiving stable therapy with other psychoactive medications, including cholinesterase inhibitors, memantine, and antidepressants, could be included if this was permitted in the study protocol.
Types of outcome measures
Primary outcomes
-
Agitation, or one or more aspects of agitation as measured by a scale that specifically measured agitation, either exclusively or as one of its components. The scales included but were not limited to:
-
-
Cohen‐Mansfield Agitation Inventory (CMAI; Cohen‐Mansfield 1986);
-
Social Dysfunction and Agitation Scale (SDAS; Wistedt 1990);
-
Clinical Global Impression Scale for Aggression (CGI; Guy 1976);
-
"Nurse Observation" scale (Colenda 1991);
-
Behavior Observation Scale of Intramural Psychogeriatric Patients (GIP; Verstraten 1988);
-
Brief Psychiatric Rating Scale (BPRS; Overall 1962; Overall 1988);
-
Overt Aggression Scale (OAS; Yudofsky 1986).
-
Secondary outcomes
-
Cognition.
-
Functional performance.
-
Overall clinical impression.
-
Effect on carers (carers' psychological morbidity or burden).
-
Incidence and severity of adverse effects.
-
Dropouts, including dropouts due to adverse events.
Search methods for identification of studies
Electronic searches
We searched ALOIS (www.medicine.ox.ac.uk/alois), which is the Cochrane Dementia and Cognitive Improvement Group's (CDCIG) Specialized Register on 2 October 2014. The search terms used were: valproic OR valproate OR divalproex.
The Information Specialists for the CDCIG maintain ALOIS, which contains studies that fall within the areas of dementia prevention, dementia treatment and management, and cognitive enhancement in healthy older populations. The studies are identified through:
-
monthly searches of a number of major healthcare databases: MEDLINE, Embase, CINAHL, PsycINFO, and Lilacs;
-
monthly searches of a number of trial registers: ISRCTN; UMIN (Japan's Trial Register); the World Health Organization (WHO) portal (which covers ClinicalTrials.gov; ISRCTN; the Chinese Clinical Trials Register; the German Clinical Trials Register; the Iranian Registry of Clinical Trials and the Netherlands National Trials Register, plus others);
-
quarterly searches of the Central Register of Controlled Trials (CENTRAL);
-
six‐monthly searches of a number of grey literature sources: ISI Web of Knowledge Conference Proceedings; Index to Theses and Australasian Digital Theses.
To view a list of all sources searched for ALOIS see About ALOIS on the ALOIS website (www.medicine.ox.ac.uk/alois).
Details of the search strategies used for the retrieval of reports of trials from the healthcare databases, CENTRAL, and conference proceedings can be viewed in the 'methods used in reviews' section within the editorial information about the Dementia and Cognitive Improvement Group.
Searching other resources
We performed additional searches in many of the sources listed above to cover the timeframe from the last searches performed for ALOIS to ensure that the search for the review was as up‐to‐date and as comprehensive as possible. The search strategies used can be seen in Appendix 1.
We carried out the most recent search for this review on 7 December 2017. Previous searches were done in October 2016, July 2010, and February 2008.
Data collection and analysis
Selection of studies
The Information Specialist of the CDCIG removed duplicates of the same references. Two review authors (ETL and JL) independently examined titles and abstracts against the prespecified inclusion criteria to exclude clearly ineligible studies. We examined any potentially eligible trial in full text. Two review authors (ETL and JL) independently evaluated full texts according to the eligibility criteria. We compared selections of trials and the review authors agreed the final list of studies. We explained final decisions for the exclusion of articles that we retrieved in full text in the Characteristics of excluded studies table.
Data extraction and management
Two review authors (ETL and JL) extracted data from each study using a data collection form that was piloted by the team. For the purpose of this updated review, the data were entered into Review Manager 5 (Review Manager 2014). Two review authors (AC and SFB) checked the data for accuracy. We also extracted data about ongoing studies, including study name, methods, participants, interventions, outcomes, starting date, contact information, and notes.
Assessment of risk of bias in included studies
Two review authors (SFB and AC) independently assessed the risk of bias in accordance with Cochrane's tool for assessing methodological quality and risk of bias (Higgins 2011). This tool assesses how the randomisation sequence was generated, how allocation was concealed, the integrity of blinding (participants, raters, and personnel), the completeness of outcome data, selective reporting, and other biases. Where inadequate details of randomisation and other characteristics of the trials were provided, we contacted authors of the studies to obtain further information.
We described the risk of bias of all included studies in the Characteristics of included studies table and narratively. In addition, we provided an overall judgement of included studies in a 'Risk of bias' summary (see Figure 1). Where the two review authors disagreed on 'Risk of bias' decisions, the final rating was made by consensus discussion involving the third member of the review team.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Measures of treatment effect
We used the mean difference (MD) to measure the treatment effect. If the same outcome was assessed using different scales, then we used the standardised mean difference (SMD). We reported 95% confidence intervals (CI). We reported results of dichotomous outcomes as odds ratios (OR) with 95% CI.
Unit of analysis issues
We considered only participant‐level outcomes. We analysed change in outcome measure from pre‐ to post‐treatment. For cross‐over trials, we used data for the first period only (if available) because of the possibility of carry‐over effects.
Dealing with missing data
To allow an intention‐to‐treat analysis, we sought data irrespective of compliance, whether or not the participant was subsequently deemed ineligible, or otherwise excluded from treatment or follow‐up. If intention‐to‐treat data were not available in the publications, we extracted 'on‐treatment' data or the data of participants who completed the trial and indicated it as such. We did not use data from titration phases prior to the randomised phase to assess safety or efficacy.
Assessment of heterogeneity
We considered clinical heterogeneity between trials (participants, interventions, and outcomes) when deciding whether or not to synthesise data. Where we performed a meta‐analysis, we used a standard Chi2 test to check for heterogeneity. We also assessed the impact of heterogeneity on the meta‐analysis using the I2 statistic.
Assessment of reporting biases
We tried to minimise the impact of publication bias by searching for both published and unpublished trials. We compared conference abstracts and registered trials with published data. We contacted the responsible organisation or the researcher for more information when we found studies in trial registries that appeared to have been completed but not published (see Description of studies). We found too few studies to allow assessment of possible publication bias using funnel plots and Egger's test for asymmetry (Egger 1997).
Data synthesis
Where data were suitable for a meta‐analysis, we presented the effect estimate from a fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
Due to the low number of included studies, subgroup analysis was not possible. Therefore, participants were combined into the category of 'dementia' regardless of subtype.
Sensitivity analysis
We did not conduct any sensitivity analyses.
'Summary of findings' table
We used the GRADE approach to assess the quality of the supporting evidence behind each estimate of treatment effect. We presented key outcomes in summary of findings Table for the main comparison, including, for each outcome, a summary of the amount of data, the magnitude of the effect size, and the overall quality of the evidence (Schünemann 2011). The measures included were: change in agitation and aggression, cognition, functional performance, and incidence and severity of adverse effects.
Results
Description of studies
Results of the search
The initial search for eligible RCTs was completed in August 2005. This identified three studies for inclusion in the review (Porsteinsson 2001; Sival 2002; Tariot 2001). An updated search on 7 February 2008 retrieved two new studies (Herrmann 2007; Tariot 2005). Further updated searches on 30 July 2010, and 4 November 2016 identified no new studies for either inclusion or exclusion in the review. The most recent search was performed in December 2017.
After removal of duplicates and first assessment by the Information Specialist of the CDCIG based on a screening of titles and abstracts, these searches resulted in a total of 41 records being passed to the authors for further scrutiny.
See Figure 2 for the flow of studies for this review.

Study flow diagram.
Included studies
We identified five studies eligible for inclusion (Herrmann 2007; Porsteinsson 2001; Sival 2002; Tariot 2001; Tariot 2005). A detailed description of each study is given in the Characteristics of included studies table.
Design
Two studies were placebo‐controlled crossover studies (Herrmann 2007; Sival 2002). In Sival 2002, there were two three‐week treatment periods separated by a one‐week washout period. In Herrmann 2007, the treatment periods lasted six weeks and there was a two‐week washout period between treatments. The remaining three studies were parallel‐group, placebo‐controlled RCTs with six‐week treatment periods (Porsteinsson 2001; Tariot 2001; Tariot 2005).
Sample size
The two crossover studies were the smallest with 14 (Herrmann 2007) and 43 (Sival 2002) participants. Porsteinsson 2001 had 56 participants, Tariot 2001 had 173, and Tariot 2005 had 153.
Setting
One study was conducted in Europe (Sival 2002), and another in Canada (Herrmann 2007). Three were multisite studies in the US (Porsteinsson 2001; Tariot 2001; Tariot 2005). All studies involved people who were institutionalised. In Sival 2002, the participants were from a short‐stay ward at a psychiatric hospital; in the other studies, participants were resident in long‐term care facilities.
Participants
See Table 1 for a description of the participants' characteristics at baseline in all studies.
| Name | Country | Population | Mean age (years) | % Women | Intervention | Diagnoses and baseline assessments | Mean MMSE |
| Canada | Multicentric, institutionalised; Alzheimer's disease | 85.6 | 42.8 | Valproate (n = 14) Placebo (n = 13) Valproate titrated to 1500 mg/day 6‐week course | AD: NINDCDS‐ADRDA criteria Agitation/aggression: CMAI | < 15 | |
| US | Multicentric, institutionalised; AD, VaD, and other dementias | 85.0 | 61.0 | Valproic acid (n = 28) Placebo (n = 28). Divalproex sodium titrated to mean 826 mg/day 6‐week course | Dementia: MMSE; DSM‐IV; NICDS‐ADRDA Agitation: CMAI Aggression: CMAI subscale Global: CGI | 6.8 | |
| The Netherlands | Institutionalised, AD, VaD, and other dementias | 80.4 | 59.5 | Valproate (n = 42) Placebo (n = 42) Sodium valproate 480 mg/day 3‐week course | Dementia: MMSE; DSM‐IV; NINCDS‐ADRDA; Clinical Dementia Rating Scale Agitation: BPRS subset Aggression: Patel's method; SDAS‐9 subscale; CGI; GIP Global: CGI | — | |
| US | Multicentric, institutionalised; AD, VaD, and other dementias | 83.3 | 64.0 | Valproic acid (n = 87) Placebo (n = 85) Divalproex sodium (delayed release); titrated to target dose of 20 mg/kg/day; median dose 1000 mg/day 6‐week course | Dementia: MMSE; DSM‐IV Agitation: CMAI Aggression: CMAI subscale Global: CGI | 7.4 | |
| US | Multicentric, institutionalised, AD | 84.0 | 68.6 | Divalproex (n = 48) Placebo (n = 78) Titrated to target dose 750 mg/day 6‐week course | Dementia (probable or possible): NINDCDS‐ADRDA Agitation, hostility, and unco‐operativeness: BPRS | 10.8 |
BPRS: Brief Psychiatric Rating Scale; CGI: Clinical Global Impression Scale; CMAI: Cohen‐Mansfield Agitation Inventory; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, 4th edition; GIP: Behavior Observation Scale of Intramural Psychogeriatric Patients; MMSE: Mini‐Mental State Examination; n: number of participants; NINDCDS‐ADRDA: National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association; SDAS‐9: 9‐item Social Dysfunction and Agitation Scale; VaD: vascular dementia.
All studies included participants with dementia, mostly with moderate‐to‐severe dementia. All studies used one or more standard methods to diagnose dementia, including Alzheimer's disease, vascular dementia, and mixed dementia (DSM‐IV (APA 1994); National Institute of Neurological and Communicative Disorders and Stroke‐Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA; McKhann 1984)).
Sival 2002 used the Clinical Dementia Rating Scale (Hughes 1982), in which 2/42 participants were classified as "light," 24/42 participants as "moderate," and 14/42 participants as "severe." This same study used the MMSE (Folstein 1975), but 14 participants could not be scored because of low level of function. All other studies also used the MMSE. Mean scores at baseline were 7.4 (Tariot 2001), 6.8 (Porsteinsson 2001), 4.5 (Herrmann 2007), and 10.8 (range 4 to 24) (Tariot 2005).
Inclusion criteria relating to degree of agitated behaviour varied between studies, but all studies required included participants to exhibit minimum levels of agitation according to standardised measures (Neuropsychiatric Inventory (NPI), BPRS, Social Dysfunction and Aggression Scale‐9).
-
Herrmann 2007 required participants to display "significant BPSD" as indicated by a score of 8 or greater on the NPI.
-
Porsteinsson 2001 required participants to have exhibited agitated behaviour for a minimum of two weeks "with sufficient intensity" to result in a BPRS score of three or more on items relating to tension, hostility, unco‐operativeness, or excitement.
-
Sival 2002 used Patel's criteria for aggressive behaviour (Patel 1993), and also required participants to score 3 or greater on at least one item of the Social Dysfunction and Aggression Scale‐9.
-
Tariot 2001 included participants who exhibited "manic symptoms" according to the Bech‐Rafaelsen Mania Scale (BRMS) and six items of the BPRS. Participants were required to score 15 or greater on the BRMS and 3 or greater on two or more of the items of the BPRS relating to tension, grandiosity, hostility, suspiciousness, unco‐operativeness, and excitement, with a total score of 15 or more.
-
Tariot 2005 required participants to have at least a two‐week history of agitation with a total score greater than 2 on the BPRS items relating to tension, hostility, unco‐operativeness, or excitement.
Interventions
Divalproex sodium
One study treated participants with divalproex sodium delayed‐release tablets or placebo (Tariot 2001). Dosage started at 125 mg twice daily and was titrated to 20 mg/kg/day to 30 mg/kg/day, to be reduced if intolerable adverse effects appeared. The median dose for treated participants at the end of six weeks was 1000 mg/day. One study treated participants with rapid‐acting divalproex sodium at an initial dose of 375 mg/day which was titrated upwards to a mean dose of 826 mg/day (Porsteinsson 2001). In this trial, a non‐blinded supervising physician, who had no contact with the blinded researchers, altered drug dosage by 125 mg/day, depending on written reports by the researchers of response and adverse effects. In Tariot 2005, participants commenced sprinkle formulation divalproex sodium 125 mg twice daily for three days, which was increased in 125 mg increments every three days to 750 mg/day or up to a maximum dose 1000 mg/day. Researchers decreased the dose by 125 mg/day if a participant experienced adverse effects. The mean dose at the end of the treatment period was 800 mg/day.
Sodium valproate
In Sival 2002, study participants received sodium valproate 240 mg twice daily for three weeks. Participants in Herrmann 2007 received valproate 125 mg liquid suspension twice daily, increased to 500 mg twice daily over the first two weeks. The dose could then be increased to a maximum of 1500 mg/day or decreased based on efficacy and tolerability as determined by a blinded study physician.
All studies permitted short‐term use of short‐acting psychotropics. Porsteinsson 2001 used chloral hydrate. Tariot 2001 permitted short‐term use of lorazepam, oxazepam, or chloral hydrate as needed. Sival 2002 allowed oxazepam for severe anxiety or insomnia. Tariot 2005 permitted zolpidem or lorazepam (or both) for severe agitation or sleep‐induction. Herrmann 2007 permitted loxapine as a rescue medication.
Outcomes
All included studies aimed to assess the effect of valproate treatments on agitation, aggression, mania, and overall function of people with dementia.
The instruments used to measure the outcomes in each study are given in Table 2.
| Outcomes | Instruments | Studies |
| Agitation and aggression | CMAI | |
| BPRS or agitation and hostility subscale, or both | ||
| BPRS | ||
| Neuropsychiatric Inventory (NPI) | ||
| Social Dysfunction and Aggression‐9 Scale (SDAS‐9) | ||
| Clinical Global Impression Scale (CGI) | ||
| Nurse Observation Scale | ||
| Patel's Method | ||
| Overt Aggression Scale | ||
| Other types of disturbed behaviour | Behavior Scale for Intramural Psychogeriatric Patients (GIP) | |
| Cognition | MMSE | |
| Functional performance | PSMS | |
| Overall clinical impression | CGI | |
| Adverse effects | Number of Adverse Reactions (checklist) | |
| Coding Symbols for Thesaurus of Adverse Reaction Terms (COSTART 1989) |
BPRS: Brief Psychiatric Rating Scale; CMAI: Cohen‐Mansfield Agitation Inventory; MMSE: Mini‐Mental State Examination; PSMS: Physical Self‐Maintenance Scale.
Agitation and aggression
The included studies used several different scales to assess change in agitated and aggressive behaviour.
Four studies used the CMAI scale to measure agitation and aggression (Herrmann 2007; Porsteinsson 2001; Tariot 2001; Tariot 2005). It was not clear which version of the CMAI was used, although three gave the same source reference for the scale (Herrmann 2007; Porsteinsson 2001; Tariot 2005). Tariot 2005 stated that they used a 36‐item version of CMAI, whereas the most commonly used version is the 29‐item version. Information obtained from the author suggested that they actually used the 29‐item version of the scale. Herrmann 2007 and Porsteinsson 2001 did not state specifically which version of the CMAI they used in their studies. We attempted to contact these authors to clarify the scale version and scoring used. Tariot 2001 used a different version of the scale specifically designed for nursing home residents (Cohen‐Mansfield 1989).
Three studies used the BPRS to assess disturbed behaviour (Porsteinsson 2001; Tariot 2001; Tariot 2005), and two studies also used the agitation and hostility subscales of the BPRS as outcome measures (Porsteinsson 2001; Tariot 2005). All three studies used the 18‐item scale but two studies rated items 0 to 6 (Tariot 2001; Tariot 2005), whereas one study rated items 1 to 7 (Porsteinsson 2001). While this would lead to disparity in total scores in each study, analysis of change in score in each study were still comparable. Information relating to the scoring of the agitation and hostility subscales of the BPRS was not available from the authors of the studies which used them, and could not be found through a further literature search.
Tariot 2001 used the BRMS to assess manic symptoms. Porsteinsson 2001 used the OAS to measure aggression. Herrmann 2007 used the NPI and its agitation subscale at their primary outcome measure. Sival 2002 used the Social Dysfunction and Aggression‐9 Scale (SDAS‐9), and the "Nurse Observation" scale, to measure incidence of aggression; the CGI to rate clinical impression of aggressive behaviour; and GIP to measure other types of disturbed behaviour.
-
The CMAI examines 29 types of agitated behaviour, including pacing, verbal or physical aggression, screaming, and restlessness. The frequency of these behaviours is measured on a 29‐item scale with each item rated from 0 (never occurs) to 6 (occurs several times an hour) and scores for physical and verbal aggression and overall aggression may be aggregated.
-
The BPRS measures physical and verbal aggression, hallucinatory behaviour, and abnormal thought content. The scale comprises 18 items each scored on a 7‐point scale with a higher score indicating higher level of dysfunction.
-
The CGI uses a 7‐point scale with scores ranging from 1 (no aggressive behaviour) to 7 (severely aggressive behaviour). It is also used to measure overall response to treatment.
-
The BRMS is an 11‐item observer‐based scale that rates the severity of manic symptoms on a 5‐point scale (Bech 1978).
-
The OAS quantifies aggressive verbal and physical behaviours and includes the number, specific nature, and intervention response.
-
The NPI is a 12‐item scale, designed to assess the severity and frequency of behavioural symptoms in people with dementia (Cummings 1994).
-
The SDAS‐9 measures several aspects of behaviour to do with patient interaction with other people, and physical and verbal aggression. The scale is a 9‐point observation scale covering outward aggressive behaviour, with total sores ranging from 0 to 36).
-
The "Nurse Observation" scale assesses the incidence of aggressive behaviour at the moment the behaviour occurs.
-
The GIP consists of 14 observational scales to describe agitated and aggressive behaviour.
Cognition
Herrmann 2007; Porsteinsson 2001; and Tariot 2005 assessed cognitive functioning using the MMSE.
Functional performance
Porsteinsson 2001 and Tariot 2005 assessed participants' functional performance using the Physical Self‐Maintenance Scale (PSMS) (Lawton 1969).
Overall clinical impression
Three studies included a rating of global clinical response of the participants using the CGI, a 7‐point scale with scores ranging from "very much improved" to "very much worse" (Porsteinsson 2001; Tariot 2001; Tariot 2005). Tariot 2001 reported this as mean change and Tariot 2005 as number of participants showing improvement. Porsteinsson 2001 reported CGI separately for therapeutic effect and adverse effects using a different Likert type scale for each.
Adverse effects
All five studies examined the tolerability, adverse effects, and safety of valproate preparations.
-
Four studies used check‐lists of adverse effects (e.g. drowsiness, nausea, vomiting, diarrhoea, confusion, disturbance in speech, disturbance of co‐ordination, tremor, seizures, oedema, fever thrombocytopenia), which were reviewed at regular intervals by interviewing participants and nursing staff; and by reviewing chart entries (Herrmann 2007; Porsteinsson 2001; Sival 2002; Tariot 2005).
-
Tariot 2001 measured adverse events based on the Coding Symbols for Thesaurus of Adverse Reaction Terms (COSTART 1989).
Effect on carers (carers psychological morbidity)
None of the included studies assessed any aspect of carer burden or well‐being.
Excluded studies
We excluded most studies on the basis of the study design (e.g. not RCTs). The studies that were excluded, with reasons for exclusion, are listed in the Characteristics of excluded studies table.
Risk of bias in included studies
See Figure 1.
Allocation
All studies included in this review indicated that participants were randomly allocated to treatments groups. However, four studies did not report the process of random sequence generation and we considered them to be at unclear risk of bias in this domain (Herrmann 2007; Porsteinsson 2001; Sival 2002; Tariot 2001). For three of these studies, there was also no specific information about allocation concealment, but Sival 2002 stated that the "code was not available to the investigators" and so we rated its risk of bias due to allocation concealment to be low. Tariot 2005 had a low risk of allocation bias.
Blinding
Two studies failed to state explicitly whether all staff involved in the study were blind to the treatment allocation of the participants (unclear risk of bias; Herrmann 2007; Tariot 2001), and two failed to state whether research staff completing the outcome measures were blinded (unclear risk of bias; Herrmann 2007; Sival 2002).
In Porsteinsson 2001, although the physicians having direct responsibility for participant care and researchers completing study assessments were blinded, a non‐blinded physician, who had no direct contact with these physicians, adjusted divalproex sodium dosage based on reports from the blinded raters. Similarly, in Sival 2002, a pharmacist and independent physician reviewed out‐of‐range laboratory results, including valproate levels. These staff had no contact with participants, investigators, the ward team or participant's relatives so we considered that the risk of introducing bias due to unblinding was low.
Tariot 2001 was described as a double‐blind study but total serum valproate levels were measured weekly and monitored by nursing staff. It was not stated whether these nursing staff were involved in the study, so we judged this to pose an unclear risk of performance bias.
Incomplete outcome data
In Tariot 2001, 54% of the valproate‐treated participants dropped out compared with 29% of control participants; 22% of all participants dropped out because of adverse effects, and the study had to be discontinued prematurely. Further, since participants had been on therapy for varying periods of time when the study was terminated, interpretation of the effects of treatment was difficult. We considered this study at high risk of attrition bias.
Porsteinsson 2001 included data in the analysis from participants who dropped out of the study, and gave the reasons for the participants dropping out (two from the divalproex group, and four from the placebo group; low risk of attrition bias).
In Tariot 2005, 11/75 participants in the divalproex group and 14/78 participants in the placebo group dropped out of treatment early, but the reasons for discontinuation were not given. However, all participants who discontinued prematurely completed final assessments which were included in the analysis, so we considered the risk of attrition bias to be low.
In Herrmann 2007, two participants dropped out during each treatment phase but reasons for this were not stated. The study authors stated that they conducted an intention‐to‐treat analysis, but data for all participants was not included in the results table. We included only the results from the first part of the study in this review and obtained first‐phase data from the study authors for all participants; for this reason, we considered the risk of attrition bias to be low.
In Sival 2002, three participants dropped out of the study due to adverse events in either the placebo or washout periods and these data were included in the analysis. One participant was excluded from the analysis due to protocol violation (low risk of attrition bias).
Selective reporting
We found no published protocols for the included studies. However, for all included studies, the results of the primary and secondary outcome measures that were specified in the methods sections of the papers were reported, as well as the frequency of adverse events. Therefore, we judged all the studies to be at low risk of selective reporting bias.
Other potential sources of bias
Sival 2002 had a cross‐over design. No results from the first phase of the study were available. The statistical analysis did not take account of the paired nature of the data ("the t‐test for independent samples is used to analyse the two‐period cross‐over trial").
We noted that three of the included studies were supported by grants from Abbott laboratories – a company which may have had a vested interest in the efficacy of the treatment – but we did not rate this as a source of bias (Porsteinsson 2001; Tariot 2001; Tariot 2005).
Effects of interventions
The included studies varied in the type of valproic acid preparation, dosage, and duration of therapy. The methods of evaluating the participants also varied between the studies, with use of different scales to assess agitation and aggression, and response to therapy. Tariot 2001 was discontinued due to the disproportionate number of dropouts in the treatment group (54%) as well as a high proportion in the placebo group (29%), with the results that not all participants received treatment for the full study period. Many of these dropouts occurred in the first three weeks (11/47 participants). Due to the high risk of attrition bias caused by the high dropout rate, we decided to exclude the data from the pooled analysis in this review. Sival 2002 was a cross‐over design and first‐phase data were not available from the published paper, or from the authors, and so data from this study was also not included in our analyses.
Agitation and aggression
We were able to pool data on agitation/aggression measured with the BPRS from two studies and the CMAI from three studies.
A meta‐analysis of agitated behaviour, assessed with total BPRS scores in two studies, showed that there was probably no difference between valproate and placebo group in total BPRS after six weeks of treatment (MD 0.23, 95% CI –2.14 to 2.59; 202 participants, 2 studies; Analysis 1.1; moderate‐quality evidence, downgraded due to imprecision) (Porsteinsson 2001; Tariot 2005). A pooled analysis of agitation measured with the agitation factor of the BPRS confirmed that there was probably no effect of treatment specifically on agitation (MD –0.67, 95% CI –1.49 to 0.15; 202 participants, 2 studies; Analysis 1.2; moderate‐quality evidence, downgraded due to imprecision). The quality of evidence on agitation measured with the CMAI was lower, but meta‐analysis of three studies that reported the change in total CMAI score between baseline and six weeks also suggested no effect on agitated behaviour (MD –1.84, 95% CI –6.02 to 2.34, 217 participants, 3 studies; I2 = 52%; Analysis 1.3; very low‐quality evidence, downgraded due to risk of bias, inconsistency, and imprecision) (Herrmann 2007; Porsteinsson 2001; Tariot 2005). Herrmann 2007 and Porsteinsson 2001 did not state specifically which version of the CMAI they used in their studies, we made the assumption that they used the standard 29‐item scale. Information from Tariot 2005 indicated that they also used the 29 item scale despite the paper stating they used a 36 item version of the scale. In light of the uncertainty regarding which version Tariot 2005 used, we repeated the pooled analysis of change in total CMAI score after excluding data from Tariot 2005 (MD 1.96, 95% CI –6.18 to 10.10; 70 participants, 2 studies; very low‐quality evidence, downgraded due to risk of bias, inconsistency, and imprecision).
Single studies only reported the other outcome measures. Porsteinsson 2001 found that there was probably little or no effect of divalproex on the hostility factor of the BPRS (MD 0.10, 95% CI –1.12 to 1.32; 55 participants, 1 study; Analysis 1.4; moderate‐quality evidence, downgraded due to imprecision) or the Overt aggression total score (MD 0.10, 95% CI –3.42 to 3.62; 55 participants, 1 study; Analysis 1.5; moderate‐quality evidence, downgraded due to imprecision). Herrmann 2007 used the NPI. This showed a clinically important difference in behavioural symptoms as measured by the NPI total score, favouring the placebo group, but there was a great deal of uncertainty about this result (MD 15.28, 95% CI ‐5.19 to 35.75; 14 participants, 1 study; Analysis 1.6; very low‐quality evidence, downgraded one level due to risk of bias and two levels due to imprecision). There was similarly a high level of uncertainty about the result on the NPI agitation/aggression subscale, which showed no clear evidence of a difference between groups (MD 1.43, 95% CI –2.48 to 5.34; 14 participants, 1 study; Analysis 1.7; very low‐quality evidence, downgraded one level due to risk of bias and two levels due to imprecision).
Cognition
Three studies assessed cognitive functioning using the MMSE (Herrmann 2007; Porsteinsson 2001; Tariot 2005). The quality of this evidence was very low. but pooled analysis of the data indicated that there may have been little or no effect of valproate on the change in MMSE score over the six‐week treatment period (MD –0.70, 95% CI –1.61 to 0.20; 217 participants, 3 studies; Analysis 1.8; very low‐quality evidence, downgraded due to risk of bias, inconsistency, and imprecision).
Functional performance
Porsteinsson 2001 and Tariot 2005 assessed functional ability using the PSMS. Pooled analysis of the change in total PSMS score indicated that there was probably little or no effect of valproate on this outcome (MD 1.19, 95% CI 0.40 to 1.98; 203 participants, 2 studies; Analysis 1.9; moderate‐quality evidence, downgraded due to imprecision).
Overall clinical impression
Three studies included a measure of global clinical change, but we excluded data from Tariot 2001 due to the very high risk of attrition bias. Tariot 2005 used the CGI as an index of clinical efficacy, measuring change in participants' overall clinical condition on a 7‐point scale (0 marked improvement to 6 marked worsening). The number of participants showing improvement was reported not to differ significantly between the two groups. Porsteinsson 2001 used the CGI to rate "therapeutic effect" on a 4‐point scale, and the presence and clinical significance of adverse effects on a 7‐point scale (from very much improved to very much worse). They reported no difference between groups in CGI ratings. Because of the different ways in which the CGI was used was used in these two studies, we were unable to pool data.
Incidence and severity of adverse effects
Meta‐analysis of three studies, all of which used divalproex sodium, found there may have been a higher rate of adverse effects among participants treated with divalproex sodium than among participants in the control group (OR 2.02, 95% CI 1.30 to 3.14; 381 participants, 3 studies; Analysis 2.27; low‐quality evidence, downgraded due to imprecision and inconsistency) (Porsteinsson 2001; Tariot 2001; Tariot 2005). A fourth study reported that the mean incidence of adverse effects was low during three weeks of observation in both sodium valproate (0.17) and placebo (0.02) groups, but the study provided no description of the types of adverse reactions or actual numbers of adverse events experienced, so we could not include this study in the meta‐analysis (Sival 2002). Data on adverse effects during the first treatment phase of Herrmann 2007 were not available in the published data or from the authors, but over the course of both treatment phases, 12 participants experienced at least one adverse event while taking valproate compared to eight participants while taking placebo. The mean number of adverse events from valproate was significantly greater than with placebo.
The descriptions of adverse effects which study authors used varied making pooled analysis of all adverse effects difficult. However, pooled analysis of adverse effects that were reported in more than one study indicated that sedation (OR 2.66, 95% CI 1.44 to 4.92; 228 participants, 2 studies; Analysis 2.1; moderate‐quality evidence, downgraded due to imprecision), 'nausea, vomiting and diarrhoea' (OR 6.92, 95% CI 2.13 to 22.49; 381 participants, 3 studies; Analysis 2.2; moderate‐quality evidence, downgraded due to imprecision), and UTIs (OR 3.07, 95% CI 1.05 to 8.97; 228 participants, 2 studies; Analysis 2.3; moderate‐quality evidence, downgraded due to imprecision) were more frequently reported among valproate‐treated participants than placebo‐treated participants. Falls, respiratory, skin or joint problems, and infections (other than UTI) were no more frequent in valproate‐treated than in placebo‐treated participants.
One study reported thrombocytopenia in 6/87 participants in the valproate group and 0/85 participants in the placebo group (Tariot 2001). One study reported thrombocytopenia in 2/14 participants during the treatment phase and none in the placebo phase (Herrmann 2007). In Porsteinsson 2001, 2/28 participants in the divalproex group had developed a significant decrease in platelet count, but not to the level of thrombocytopenia. Sival 2002 monitored blood counts, but reported no instances of a drop in platelet count.
Serious adverse events
The included studies varied in the reporting of serious adverse events (SAE) during their treatment period. Sival 2002 did not report the incidence of SAE during their study. Herrmann 2007 did not report numbers of participants who experienced SAE clearly, but the study stated that two participants in the treatment phase had falls that rated as SAEs. There was no indication given about whether these falls were considered related to the study medication and it was not clear whether these were the only SAEs to occur. Tariot 2005 also did not report specifically on numbers of SAEs although there was one death in the drug‐treatment group which was not considered related to the study drug. The authors stated that most adverse events were rated as mild to moderate in severity and were judged as not related to the study drug. Porsteinsson 2001 reported four SAEs, one in the placebo group (worsening of chronic renal failure) and three in the divalproex group (one with seizure, cerebrovascular accident, and pneumonia; one with seizure; and one with small bowel obstruction). Tariot 2001 reported one SAE due to hyponatraemia in the divalproex group, which was thought probably to be related to the study drug. Six other participants experienced SAEs, five in the divalproex group and one in the placebo group. These SAEs were four hospitalisations (for cellulitis, dehydration, pneumonia, myocardial infarction, and constipation) and one cerebrovascular accident; all were considered to be unrelated to the study drug.
Pooled analysis of the number of SAEs for the two studies which did report data indicated that participants treated with valproate were more likely to experience SAEs (OR 4.77, 95% CI 1.00 to 22.74; 228 participants, 2 studies; Analysis 2.28; very low‐quality evidence, downgraded due to risk of bias, inconsistency, and imprecision) (Porsteinsson 2001; Tariot 2001).
Dropouts
All included studies reported that there were participants who dropped out during the study period. For most studies, the number of dropouts were not disproportionate between the treatment and the placebo groups (Porsteinsson 2001: 7% divalproex versus 14% placebo; Sival 2002: 0% valproate versus 5% placebo; Tariot 2005: 15% divalproex versus 18% placebo; Herrmann 2007: 7% valproate versus 7% placebo). However, in the study by Tariot 2001, a disproportionate number of participants in the treatment group dropped out of the study (54% divalproex versus 29% placebo), with 22% from the treatment group compared to only 4% of the placebo group withdrawing due to adverse events. These adverse events were predominantly related to somnolence but also included hyponatraemia, accidents, and weight loss. Due to this disproportionate level of withdrawal in the divalproex group, the study was terminated early.
Discussion
Summary of main results
We found no evidence of a beneficial effect of valproate on our primary outcome of agitation or closely related behavioural outcomes, measured using several outcome scales. Neither did we find evidence of any important effect on any of our secondary efficacy outcomes. However, participants taking valproate may have been at higher risk of adverse effects, including SAEs. These findings are described in summary of findings Table for the main comparison.
Specifically, our results showed that there was probably little or no effect on agitated and aggressive behaviours as measured by the BPRS. There was very low‐quality evidence using other scales, but results were consistent with this finding. Pooled analysis also indicated probably slightly worse function in the valproate‐treated group, on functional ability assessed with the PSMS, of uncertain clinical importance. We found very low‐quality evidence of no effect on cognition assessed with the MMSE.
Pooled analysis of the numbers of participants who experienced any adverse effect was limited to three of the included studies (Porsteinsson 2001; Tariot 2001; Tariot 2005), but indicated that participants treated with valproate preparations may have been more likely to experience adverse effects than participants taking placebo. Likewise, participants treated with valproate preparations were more likely to experience SAEs, although the quality of this evidence was very low (data from two studies; Porsteinsson 2001; Tariot 2001). Individual adverse effects which were found in one or more studies to be more frequent in the valproate‐treated group were sedation, gastrointestinal symptoms, and UTI.
Overall completeness and applicability of evidence
The small number of included studies, some of which involved small numbers of participants, limited the evidence available. This was further limited since one study closed prematurely and that separate first‐phase data were not available from the cross‐over trials for inclusion in the review analysis (Herrmann 2007 for some data; Sival 2002).
Because of the limited number of participants, it was not possible to analyse secondary objectives such as the effect of valproate therapy on individual manifestations of agitation (e.g. crying out, wandering) or the influence of age, gender, or degree and type of dementia on the response to therapy. The small number of included studies also meant it was not possible to analyse how the response to valproate preparations was influenced by dose and duration of treatment.
The premature termination of one study, in which 47 (54%) treated participants dropped out before the protocol could be completed, severely limited the confidence that could be placed on the conclusion of the study authors that divalproex sodium improved agitation of people with dementia (Tariot 2001). Because so many of the participants did not complete the study treatment period, we did not include data from this study in the pooled efficacy analyses.
Quality of the evidence
We only identified five studies for inclusion in this review and most varied in the outcome measures used to assess impact of treatment with valproate preparations on agitated behaviour thus making comparisons of study outcomes difficult. We found the quality of evidence for most outcomes measures used to be of low or very low quality primarily due to risk of bias, imprecision, and inconsistency in the included studies (because of poorly described methodology, small sample sizes, and heterogeneity of sample groups and treatments used).
Methodological and clinical diversity limited opportunities for pooling data. Specifically, variations in method, type of medication, dosage, duration of treatment, and use of different outcome measures in these studies made it difficult to apply meta‐analysis. For example, it was difficult to compare directly studies that employed short‐acting sodium valproate (Sival 2002) or longer‐acting divalproex sodium (Porsteinsson 2001; Tariot 2001), and in which the dosage varied more than two‐fold (Sival 2002, mean dose 480 mg/day; Tariot 2001, median dose 1000 mg/day; Porsteinsson 2001, mean dose 875 mg/day).
We did not include data from Sival 2002 because we could not obtain first period data as our protocol required. We considered that there was a risk of carryover effects. We also considered that there was a unit of analysis error in the analysis reported in the paper, which failed to account for the cross‐over design.
Potential biases in the review process
It is possible that pooling of clinically diverse studies may have concealed important benefits or harms.
Agreements and disagreements with other studies or reviews
The conclusions of this updated review were in keeping with the NICE evidence summary on the use of valproate preparations for agitation and aggression in dementia which stated that such medications were no more effective than placebo, and that adverse effects were more common in people taking them (NICE 2015).

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Study flow diagram.

Comparison 1 Valproate preparations versus placebo, Outcome 1 Brief Psychiatric Rating Scale (BPRS) total score. Change from baseline at 6 weeks (intention to treat (ITT)).

Comparison 1 Valproate preparations versus placebo, Outcome 2 BPRS agitation factor. Change from baseline at 6 weeks (ITT).

Comparison 1 Valproate preparations versus placebo, Outcome 3 Cohen‐Mansfield Agitation Index. Total Score. Change from baseline at 6 weeks (ITT).

Comparison 1 Valproate preparations versus placebo, Outcome 4 BPRS hostility factor. Change from baseline at 6 weeks (ITT).

Comparison 1 Valproate preparations versus placebo, Outcome 5 Overt Aggression Scale total score. Change from baseline at 6 weeks (ITT).
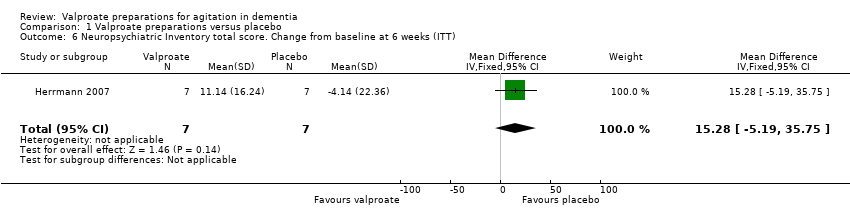
Comparison 1 Valproate preparations versus placebo, Outcome 6 Neuropsychiatric Inventory total score. Change from baseline at 6 weeks (ITT).

Comparison 1 Valproate preparations versus placebo, Outcome 7 Neuropsychiatric Inventory Agitation/Aggression subscore. Change from baseline at 6 weeks (ITT).

Comparison 1 Valproate preparations versus placebo, Outcome 8 Mini‐Mental State Examination total score. Change from baseline at 6 weeks (ITT).

Comparison 1 Valproate preparations versus placebo, Outcome 9 Physical Self‐Maintenance Scale total score. Change from baseline at 6 weeks (ITT).
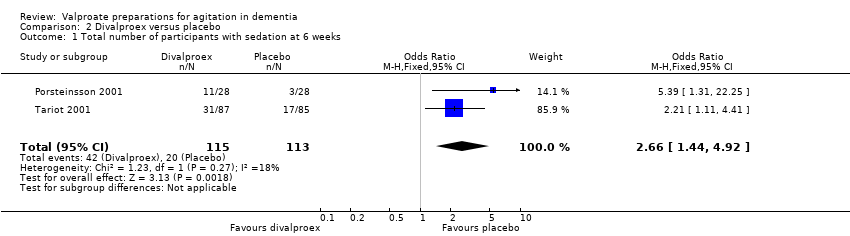
Comparison 2 Divalproex versus placebo, Outcome 1 Total number of participants with sedation at 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 2 Total number of participants with nausea, vomiting, or diarrhoea at 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 3 Total number of participants with a urinary tract infection by 6 weeks.
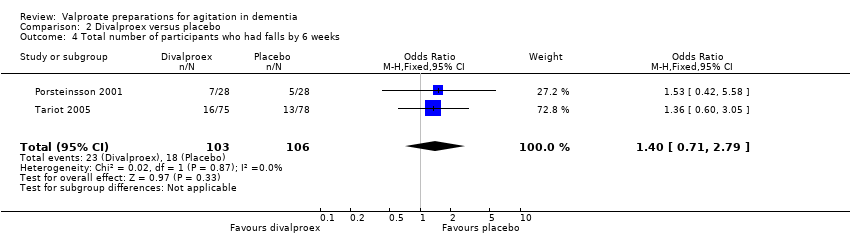
Comparison 2 Divalproex versus placebo, Outcome 4 Total number of participants who had falls by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 5 Total number of participants with general disorders by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 6 Total number of participants with postural instability by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 7 Total number of participants with weakness by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 8 Total number of participants with cardiovascular problems by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 9 Total number of participants with oedema by 6 weeks.
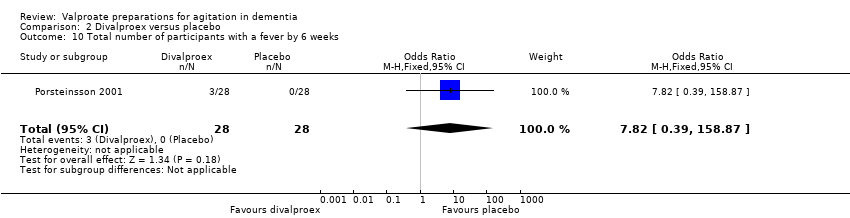
Comparison 2 Divalproex versus placebo, Outcome 10 Total number of participants with a fever by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 11 Total number of participants with a respiratory problem by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 12 Total number of participants with ataxia at 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 13 Total number of participants with a skin problem at 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 14 Total number of participants with trauma (other than falls) by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 15 Total number of participants with thrombocytopenia by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 16 Total number of participants with joint problems by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 17 Total number of participants with other infection by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 18 Total number of participants with hallucinations by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 19 Total number of participants with accidental injury by 6 weeks.
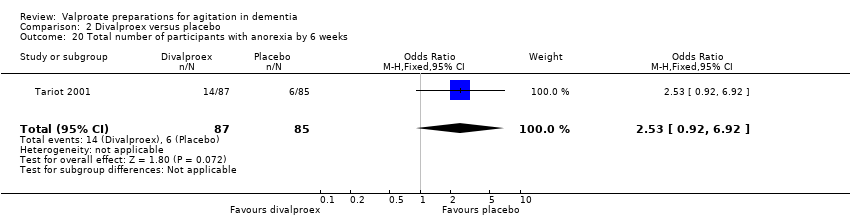
Comparison 2 Divalproex versus placebo, Outcome 20 Total number of participants with anorexia by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 21 Total number of participants with weight loss by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 22 Total number of participants with dehydration by 6 weeks.
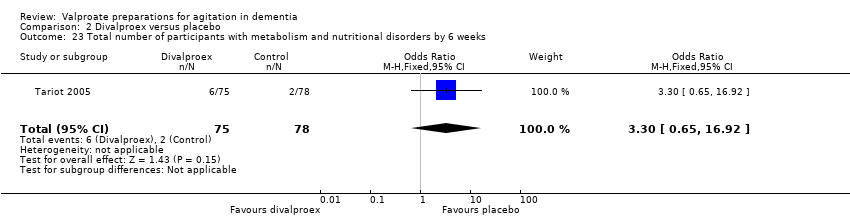
Comparison 2 Divalproex versus placebo, Outcome 23 Total number of participants with metabolism and nutritional disorders by 6 weeks.
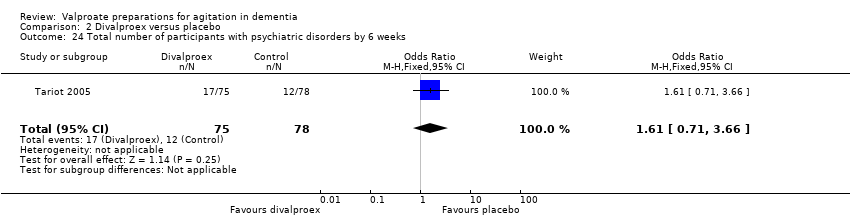
Comparison 2 Divalproex versus placebo, Outcome 24 Total number of participants with psychiatric disorders by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 25 Total number of participants with other gastrointestinal problem by 6 weeks.
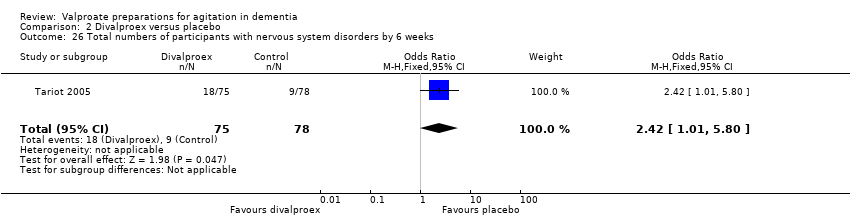
Comparison 2 Divalproex versus placebo, Outcome 26 Total numbers of participants with nervous system disorders by 6 weeks.
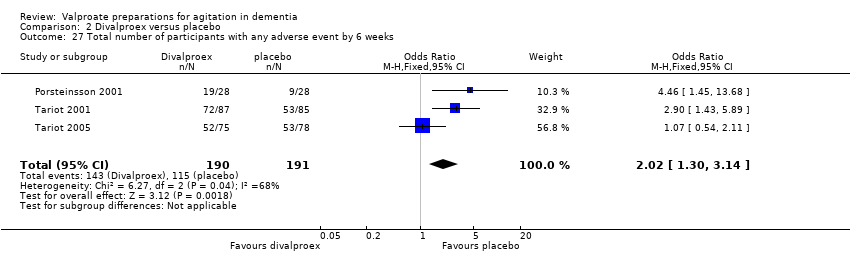
Comparison 2 Divalproex versus placebo, Outcome 27 Total number of participants with any adverse event by 6 weeks.

Comparison 2 Divalproex versus placebo, Outcome 28 Total number of participants with serious adverse events by 6 weeks.
| Valproate preparations compared to placebo for agitation in dementia | ||||||
| Patient or population: people with agitation in dementia | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with valproate preparations | |||||
| Agitation and aggression Scale: 0–108 (higher score indicated higher level of dysfunction) | The mean change from baseline for agitation and aggression was –5.34 points. | MD 0.23 higher | — | 202 | ⊕⊕⊕⊝ | — |
| Agitation and aggression Scale: 0–18 (higher score indicated higher level of dysfunction) | The mean change from baseline for agitation and aggression was –1.88 points. | MD 0.67 lower | — | 202 | ⊕⊕⊕⊝ | — |
| Agitation and aggression Scale: 0–216 (higher score indicated more agitated behaviour) | The mean change from baseline for agitation and aggression was –4.42 points. | MD 1.84 lower | — | 217 | ⊕⊝⊝⊝ | — |
| Cognition Scale: 0–30 (lower score indicated greater cognitive impairment) | The mean change from baseline for cognition was 0.46 points. | MD 0.7 lower | — | 217 | ⊕⊝⊝⊝ | — |
| Functional performance Scale: 6–30 (higher score indicated greater impairment in ADL) | The mean change from baseline for functional performance was 0.06 points. | MD 1.19 higher | — | 203 | ⊕⊕⊕⊝ | — |
| Any adverse event by 6 weeks | Study population | OR 2.02 | 381 | ⊕⊕⊝⊝ | — | |
| 602 per 1000 | 753 per 1000 | |||||
| Serious adverse events by 6 weeks | Study population | OR 4.77 | 228 | ⊕⊝⊝⊝ | — | |
| 18 per 1000 | 79 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ADL: activities of daily living; CI: confidence interval; ITT: intention to treat; MD: mean difference; OR: odds ratio; RCT: randomised controlled trial. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level for imprecision due to small number of participants. bDowngraded in light of imprecision due to confidence intervals including the potential for harm or benefit. cDowngraded one level due to inconsistency (heterogeneity between studies). dDowngraded one level due to study limitations (risk of bias). | ||||||
| Name | Country | Population | Mean age (years) | % Women | Intervention | Diagnoses and baseline assessments | Mean MMSE |
| Canada | Multicentric, institutionalised; Alzheimer's disease | 85.6 | 42.8 | Valproate (n = 14) Placebo (n = 13) Valproate titrated to 1500 mg/day 6‐week course | AD: NINDCDS‐ADRDA criteria Agitation/aggression: CMAI | < 15 | |
| US | Multicentric, institutionalised; AD, VaD, and other dementias | 85.0 | 61.0 | Valproic acid (n = 28) Placebo (n = 28). Divalproex sodium titrated to mean 826 mg/day 6‐week course | Dementia: MMSE; DSM‐IV; NICDS‐ADRDA Agitation: CMAI Aggression: CMAI subscale Global: CGI | 6.8 | |
| The Netherlands | Institutionalised, AD, VaD, and other dementias | 80.4 | 59.5 | Valproate (n = 42) Placebo (n = 42) Sodium valproate 480 mg/day 3‐week course | Dementia: MMSE; DSM‐IV; NINCDS‐ADRDA; Clinical Dementia Rating Scale Agitation: BPRS subset Aggression: Patel's method; SDAS‐9 subscale; CGI; GIP Global: CGI | — | |
| US | Multicentric, institutionalised; AD, VaD, and other dementias | 83.3 | 64.0 | Valproic acid (n = 87) Placebo (n = 85) Divalproex sodium (delayed release); titrated to target dose of 20 mg/kg/day; median dose 1000 mg/day 6‐week course | Dementia: MMSE; DSM‐IV Agitation: CMAI Aggression: CMAI subscale Global: CGI | 7.4 | |
| US | Multicentric, institutionalised, AD | 84.0 | 68.6 | Divalproex (n = 48) Placebo (n = 78) Titrated to target dose 750 mg/day 6‐week course | Dementia (probable or possible): NINDCDS‐ADRDA Agitation, hostility, and unco‐operativeness: BPRS | 10.8 | |
| BPRS: Brief Psychiatric Rating Scale; CGI: Clinical Global Impression Scale; CMAI: Cohen‐Mansfield Agitation Inventory; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, 4th edition; GIP: Behavior Observation Scale of Intramural Psychogeriatric Patients; MMSE: Mini‐Mental State Examination; n: number of participants; NINDCDS‐ADRDA: National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association; SDAS‐9: 9‐item Social Dysfunction and Agitation Scale; VaD: vascular dementia. | |||||||
| Outcomes | Instruments | Studies |
| Agitation and aggression | CMAI | |
| BPRS or agitation and hostility subscale, or both | ||
| BPRS | ||
| Neuropsychiatric Inventory (NPI) | ||
| Social Dysfunction and Aggression‐9 Scale (SDAS‐9) | ||
| Clinical Global Impression Scale (CGI) | ||
| Nurse Observation Scale | ||
| Patel's Method | ||
| Overt Aggression Scale | ||
| Other types of disturbed behaviour | Behavior Scale for Intramural Psychogeriatric Patients (GIP) | |
| Cognition | MMSE | |
| Functional performance | PSMS | |
| Overall clinical impression | CGI | |
| Adverse effects | Number of Adverse Reactions (checklist) | |
| Coding Symbols for Thesaurus of Adverse Reaction Terms (COSTART 1989) | ||
| BPRS: Brief Psychiatric Rating Scale; CMAI: Cohen‐Mansfield Agitation Inventory; MMSE: Mini‐Mental State Examination; PSMS: Physical Self‐Maintenance Scale. | ||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Brief Psychiatric Rating Scale (BPRS) total score. Change from baseline at 6 weeks (intention to treat (ITT)) Show forest plot | 2 | 203 | Mean Difference (IV, Fixed, 95% CI) | 0.23 [‐2.14, 2.59] |
| 2 BPRS agitation factor. Change from baseline at 6 weeks (ITT) Show forest plot | 2 | 203 | Mean Difference (IV, Fixed, 95% CI) | ‐0.67 [‐1.49, 0.15] |
| 3 Cohen‐Mansfield Agitation Index. Total Score. Change from baseline at 6 weeks (ITT) Show forest plot | 3 | 217 | Mean Difference (IV, Fixed, 95% CI) | ‐1.84 [‐6.02, 2.34] |
| 4 BPRS hostility factor. Change from baseline at 6 weeks (ITT) Show forest plot | 1 | 55 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐1.12, 1.32] |
| 5 Overt Aggression Scale total score. Change from baseline at 6 weeks (ITT) Show forest plot | 1 | 55 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐3.42, 3.62] |
| 6 Neuropsychiatric Inventory total score. Change from baseline at 6 weeks (ITT) Show forest plot | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 15.28 [‐5.19, 35.75] |
| 7 Neuropsychiatric Inventory Agitation/Aggression subscore. Change from baseline at 6 weeks (ITT) Show forest plot | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | 1.43 [‐2.48, 5.34] |
| 8 Mini‐Mental State Examination total score. Change from baseline at 6 weeks (ITT) Show forest plot | 3 | 217 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70 [‐1.61, 0.20] |
| 9 Physical Self‐Maintenance Scale total score. Change from baseline at 6 weeks (ITT) Show forest plot | 2 | 203 | Mean Difference (IV, Fixed, 95% CI) | 1.19 [0.40, 1.98] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Total number of participants with sedation at 6 weeks Show forest plot | 2 | 228 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.66 [1.44, 4.92] |
| 2 Total number of participants with nausea, vomiting, or diarrhoea at 6 weeks Show forest plot | 3 | 381 | Odds Ratio (M‐H, Fixed, 95% CI) | 6.92 [2.13, 22.49] |
| 3 Total number of participants with a urinary tract infection by 6 weeks Show forest plot | 2 | 228 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.07 [1.05, 8.97] |
| 4 Total number of participants who had falls by 6 weeks Show forest plot | 2 | 209 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.71, 2.79] |
| 5 Total number of participants with general disorders by 6 weeks Show forest plot | 1 | 153 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.14 [1.05, 4.36] |
| 6 Total number of participants with postural instability by 6 weeks Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.5 [0.47, 43.09] |
| 7 Total number of participants with weakness by 6 weeks Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 10.47 [0.54, 204.32] |
| 8 Total number of participants with cardiovascular problems by 6 weeks Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.08 [0.18, 24.31] |
| 9 Total number of participants with oedema by 6 weeks Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 7.82 [0.39, 158.87] |
| 10 Total number of participants with a fever by 6 weeks Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 7.82 [0.39, 158.87] |
| 11 Total number of participants with a respiratory problem by 6 weeks Show forest plot | 2 | 209 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.33, 2.46] |
| 12 Total number of participants with ataxia at 6 weeks Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.28, 6.87] |
| 13 Total number of participants with a skin problem at 6 weeks Show forest plot | 3 | 381 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.55, 1.91] |
| 14 Total number of participants with trauma (other than falls) by 6 weeks Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.10, 4.17] |
| 15 Total number of participants with thrombocytopenia by 6 weeks Show forest plot | 1 | 172 | Odds Ratio (M‐H, Fixed, 95% CI) | 13.64 [0.76, 245.98] |
| 16 Total number of participants with joint problems by 6 weeks Show forest plot | 2 | 209 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.35, 2.05] |
| 17 Total number of participants with other infection by 6 weeks Show forest plot | 3 | 381 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.70, 2.45] |
| 18 Total number of participants with hallucinations by 6 weeks Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.11 [0.12, 79.64] |
| 19 Total number of participants with accidental injury by 6 weeks Show forest plot | 2 | 325 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.84, 2.50] |
| 20 Total number of participants with anorexia by 6 weeks Show forest plot | 1 | 172 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.53 [0.92, 6.92] |
| 21 Total number of participants with weight loss by 6 weeks Show forest plot | 1 | 172 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.77 [0.71, 10.81] |
| 22 Total number of participants with dehydration by 6 weeks Show forest plot | 1 | 172 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.63 [0.73, 18.01] |
| 23 Total number of participants with metabolism and nutritional disorders by 6 weeks Show forest plot | 1 | 153 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.30 [0.65, 16.92] |
| 24 Total number of participants with psychiatric disorders by 6 weeks Show forest plot | 1 | 153 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.71, 3.66] |
| 25 Total number of participants with other gastrointestinal problem by 6 weeks Show forest plot | 2 | 208 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.70, 2.97] |
| 26 Total numbers of participants with nervous system disorders by 6 weeks Show forest plot | 1 | 153 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.42 [1.01, 5.80] |
| 27 Total number of participants with any adverse event by 6 weeks Show forest plot | 3 | 381 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.02 [1.30, 3.14] |
| 28 Total number of participants with serious adverse events by 6 weeks Show forest plot | 2 | 228 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.77 [1.00, 22.74] |