Quimioterapia en dosis elevadas y trasplante de médula ósea o células tronco autólogas versus quimioterapia convencional para las mujeres con cáncer de mama de mal pronóstico temprano
Resumen
Antecedentes
Las tasas de supervivencia general son decepcionantes para las mujeres con cáncer de mama de mal pronóstico temprano. El trasplante autólogo de médula ósea o de células madre periféricas (en el que la mujer es a la vez donante y receptora) se ha considerado una técnica prometedora porque permite utilizar dosis mucho más altas de quimioterapia.
Objetivos
Comparar la eficacia y la seguridad de la quimioterapia en dosis elevadas y el autoinjerto (sea trasplante de médula ósea o de células madre autólogas) con quimioterapia convencional para las mujeres con cáncer de mama de mal pronóstico temprano.
Métodos de búsqueda
Se hicieron búsquedas en el registro especializado del Grupo Cochrane de Cáncer de Mama (Cochrane Breast Cancer Group Specialised Register), MEDLINE (1966 a octubre de 2015), EMBASE (1980 a octubre de 2015), la International Clinical Trials Registry Search Platform de la Organización Mundial de la Salud y en ClinicalTrials.gov el 21 de octubre de 2015.
Criterios de selección
Ensayos controlados aleatorizados (ECA) que comparan la quimioterapia de alta dosis y el autoinjerto (trasplante de médula ósea o rescate de células madre) versus la quimioterapia sin autoinjerto para mujeres con cáncer de mama temprano de mal pronóstico.
Obtención y análisis de los datos
Dos autores de la revisión seleccionaron los ECA, extrajeron los datos y evaluaron el riesgo de sesgo de forma independiente. Se combinaron los datos mediante un modelo de efectos fijos de Mantel‐Haenszel para calcular los riesgos relativos (RR) agrupados y los intervalos de confianza (IC) del 95%. La calidad de la evidencia se evaluó mediante los métodos GRADE. Los resultados fueron las tasas de supervivencia, la toxicidad y la calidad de vida.
Resultados principales
Se incluyeron 14 ECA de 5600 mujeres asignadas al azar a recibir quimioterapia de alta dosis y autoinjerto (trasplante de médula ósea o rescate de células madre) versus quimioterapia sin autoinjerto para mujeres con cáncer de mama temprano de mal pronóstico. Los ensayos presentaron un bajo riesgo de sesgo en la mayoría de áreas.
Existe evidencia de alta calidad de que la quimioterapia de alta dosis no aumenta la probabilidad de supervivencia general en ninguna etapa del seguimiento (a los tres años: RR 1,02, IC 95% 0,95 a 1,10, 3 ECA, 795 mujeres, I² = 56%; a los cinco años: RR 1,00, IC 95% 0,96 a 1,04, 9 ECA, 3948 mujeres, I² = 0%; a los seis años: RR 0,94, IC del 95%: 0,81 a 1,08, 1 ECA, 511 mujeres; a los ocho años: RR1,17, IC del 95%: 0,95 a 1,43, 1 ECA, 344 mujeres; a los 12 años: RR 1,18; IC del 95%: 0,99 a 1,42, 1; un ECA, 382 mujeres).
Hay evidencia de alta calidad de que la quimioterapia en dosis altas mejora la probabilidad de supervivencia sin eventos a los tres años (RR 1,19, IC del 95%: 1,06 a 1,34, 3 ECA, 795 mujeres, I² = 56%), pero este efecto ya no fue evidente a una duración más larga del seguimiento (a los cinco años: RR 1,04, IC del 95%: 0,99 a 1,09, 9 ECA, 3948 mujeres, I² = 14%; a los seis años RR 1,04, IC del 95%: 0,87 a 1,24, 1 ECA, 511 mujeres; a los ocho años: RR 1,27, IC del 95%: 0,99 a 1,64, 1 ECA, 344 mujeres; a los 12 años: RR 1,18, IC del 95%: 0,95 a 1,45, 1 ECA, 382 mujeres).
Las muertes relacionadas con el tratamiento fueron mucho más frecuentes en el grupo de dosis altas (RR 7,97, IC del 95%: 3,99 a 15,92, 14 ECA, 5600 mujeres, I² = 12%, evidencia de alta calidad) y la morbilidad no mortal también fue más frecuente y más grave en el grupo de dosis altas. Hubo poca o ninguna diferencia entre los grupos en la incidencia de segundos cánceres a los cuatro a nueve años de mediana de seguimiento (RR 1,25; IC del 95%: 0,90 a 1,73, 7 ECA, 3423 mujeres, I² = 0%, evidencia de alta calidad). Las mujeres del grupo de dosis altas informaron de puntuaciones de calidad de vida significativamente peores inmediatamente después del tratamiento, pero hubo pocas diferencias estadísticamente significativas entre los grupos al año.
Los estudios primarios tuvieron un bajo riesgo de sesgo en la mayoría de las áreas, y las evidencia se evaluó mediante los métodos de GRADE y se calificó como de alta calidad en todas las comparaciones.
Conclusiones de los autores
Existe evidencia de alta calidad de que el uso de altas dosis de quimioterapia con autoinjerto en mujeres con cáncer de mama de mal pronóstico temprano conlleva un aumento de la mortalidad relacionada con el tratamiento y poco o ningún aumento de la supervivencia.
PICOs
Resumen en términos sencillos
Altas dosis de quimioterapia y trasplante de médula ósea o de células madre para el cáncer de mama con mal pronóstico temprano utilizando las células propias de la mujer (autólogas)
Antecedentes
Las mujeres con cáncer de mama que tienen múltiples ganglios linfáticos positivos cuando se les diagnostica por primera vez tienen un alto riesgo de recurrencia. La quimioterapia convencional tiene un éxito limitado y no es segura a altas dosis ya que daña la médula ósea. Un tratamiento considerado prometedor consistía en administrar a las mujeres dosis muy altas de quimioterapia, seguidas de un trasplante de células madre para regenerar su médula ósea. Los autores de la revisión Cochrane examinaron la evidencia actualizada hasta octubre de 2015.
Características de los estudios
Se incluyeron 14 ensayos controlados aleatorizados (5600 mujeres) que compararon la quimioterapia de alta dosis versus la quimioterapia convencional en mujeres con cáncer de mama temprano y con un alto riesgo de recidiva. Se definieron como mujeres con cáncer de mama que se ha propagado a múltiples ganglios linfáticos locales sin ninguna evidencia de propagación más allá de los ganglios linfáticos locales.
Todos los estudios informaron sobre su fuente de financiación. Ocho estudios fueron financiados por organizaciones sin ánimo de lucro, uno por una compañía pública de seguros de salud, uno por fuentes de la industria y cuatro por una combinación de organizaciones sin ánimo de lucro y fuentes de la industria. Cuatro de los estudios informaron de que los autores no tenían ningún conflicto de intereses potencial, seis informaron de que uno o más de sus autores habían recibido algún tipo de apoyo de las empresas farmacéuticas, y cuatro no mencionaron si alguno de sus autores tenía algún conflicto de intereses potencial.
Resultados clave
El uso de altas dosis de quimioterapia tiene poco o ningún efecto en el aumento de la supervivencia. Aunque las tasas de supervivencia sin incidentes fueron más altas en el brazo de dosis altas durante el seguimiento de tres años, este efecto no fue evidente en el seguimiento más largo. Las muertes relacionadas con el tratamiento eran mucho más habituales en el grupo de dosis altas. Los efectos secundarios también fueron más comunes y más severos en el grupo de dosis altas. No se encontró un efecto en el número de mujeres que desarrollan un segundo cáncer.
Calidad de la evidencia
La evidencia fue de alta calidad.
Authors' conclusions
Summary of findings
| High‐dose chemotherapy versus chemotherapy without bone marrow transplant or stem cell rescue | ||||||
| Population: women with early poor prognosis breast cancer | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with standard chemotherapy | Risk with high dose chemotherapy | |||||
| Overall survival at 5‐year follow‐up | 672 per 1000 | 672 per 1000 | RR 1.00 | 3566 (8 RCTs) | ⨁⨁⨁⨁ | |
| Event‐free survival at 5‐year follow‐up | 578 per 1000 | 601 per 1000 | RR 1.04 | 3566 | ⨁⨁⨁⨁ | |
| Treatment‐related mortality | 2 per 1000 | 14 per 1000 | RR 7.97 | 5600 | ⨁⨁⨁⨁ | Most deaths occurred within the first year of treatment |
| Second cancers at 4 ‐ 9‐year median follow‐up | 25 per 1000 | 31 per 1000 | RR 1.25 | 3423 | ⨁⨁⨁⨁ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the median risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
Background
Description of the condition
Breast cancer is the most common cancer occurring in women and is the primary cause of cancer deaths among women worldwide (Ferlay 2015; WHO 2000). Its incidence is increasing in most countries (Bray 2004). The lifetime risk of a woman developing breast cancer is about one in eight in the United States and one in nine in England and Wales (ACS 2002; DOH 2002).
In women with early breast cancer, all detectable cancer is restricted to the breast or local lymph nodes (Clarke 2008). Women who have multiple positive lymph nodes when they are first diagnosed are at high risk of recurrent disease. Without adjuvant chemotherapy, the median recurrence rate at five years is over 60% for women with four to nine positive nodes and over 70% for women with more than 10 positive nodes (Nemoto 1980). Established chemotherapy regimens have a high failure rate, improving the chance of 10‐year survival by about 11% for women under 50 and about 3% for older women (Clarke 2008).
Description of the intervention
Researchers from the 1970s onwards described a dose‐response relationship in the action of chemotherapy drugs against cancer (Frei 1980); thus it was observed that in the treatment of breast cancer the percentage of women responding to therapy is positively associated with the dose intensity of the drugs received, dose intensity being a function of both the dose and the timing of the chemotherapy regimen (Hryniuk 1986). The technique of autologous bone marrow or peripheral blood stem cell transplant (autograft) was considered an exciting development because it addressed the problem of bone‐marrow toxicity which had previously limited the dose of chemotherapy drugs that could be safely given. The use of autograft with chemotherapy permitted the administration of doses many times higher than could otherwise be used. Results of animal studies were encouraging, as were non‐randomised patient trials which commenced during the 1980s and which achieved prolonged survival times using high‐dose chemotherapy and autograft for women with advanced breast cancer (Antman 1992; Peters 1988; Williams 1992).
Why it is important to do this review
Evidence from non‐randomised studies has been criticised for participant selection bias and other design weaknesses (Eddy 1992). The first randomised trials began in 1991. Several randomised controlled trials have now been carried out among women with early poor prognosis breast cancer, which is defined as breast cancer that has spread to multiple local lymph nodes without any evidence of distant metastases (spread). The aim of this review was to consider these studies with respect to the relative effectiveness and safety of the treatments they compare.
Objectives
To compare the effectiveness and safety of high dose chemotherapy and autograft (either autologous bone marrow or stem cell transplantation) with conventional chemotherapy for women with early poor prognosis breast cancer.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials were eligible for the review.
Types of participants
Women of any age with early poor prognosis breast cancer either at first diagnosis or as a recurrence, whether or not previously treated with chemotherapy. We include women with any size of breast tumour. We define 'early poor prognosis breast cancer' in this review as breast cancer that has spread to multiple local lymph nodes without any evidence of distant metastases
Types of interventions
High‐dose chemotherapy with autologous bone marrow or stem cell transplantation versus conventional chemotherapy, regardless of the duration of therapy. We define 'high‐dose chemotherapy' as chemotherapy sufficient to require bone marrow transplantation or stem cell rescue. We define 'conventional therapy' as chemotherapy at a lower dose than the high‐dose therapy and without bone marrow transplant or stem cell rescue.
Types of outcome measures
Primary outcomes
-
Overall survival (measured at 3, 4, 5, 6 or more years)
-
Event‐free survival (no evidence of recurrence of breast cancer: measured at 3, 4, 5, 6 or more years)
Secondary outcomes
-
Treatment‐related mortality
-
Morbidity such as non‐haematological toxicities, e.g. nausea and vomiting, white cell measures, new malignancies
-
Quality‐of‐life measures
Search methods for identification of studies
Electronic searches
We searched the following databases on 21 October 2015:
-
The Cochrane Breast Cancer Group's (CBCG's) Specialised Register. Details of search strategies used by the CBCG for the identification of studies and the procedures used to code references are outlined in the CBCG's module at www.mrw.interscience.wiley.com/cochrane/clabout/articles/BREASTCA/frame.html. We retrieved trials with the text‐words bone marrow transplantation, stem cell transplantation, stem cell support, autologous stem cell support, high dose chemotherapy, and chemotherapy.
-
MEDLINE (via OvidSP) until October 2015 (See Appendix 1).
-
EMBASE (via Embase.com) until October 2015 (See Appendix 2).
-
The World Health Organization's (WHO) International Clinical Trials Registry Platform (ICTRP) search portal (apps.who.int/trialsearch/Default.aspx) for prospectively‐registered and ongoing trials until October 2015 (See Appendix 3).
-
ClinicalTrials.gov (clinicaltrials.gov/ct2/search/advanced) until October 2015 (See Appendix 4).
Searching other resources
We searched the reference lists of articles found by the above search strategy.
Data collection and analysis
Selection of studies
For the 2015 review update, two review authors (JM and MA or AL) undertook study selection, and independently screened all articles retrieved by the search. Three review authors (JM, MA or AL and CF) independently assessed whether the studies met the inclusion criteria, with disagreements resolved by discussion.
For previous versions of the review, three review authors (CF, RB and JMB), one of whom (RB) is a content expert, had undertaken study selection. CF screened the titles and abstracts of articles found in the search, and discarded studies that were clearly ineligible; however, the aim was to be overly inclusive rather than risk losing relevant studies. CF obtained copies of the full‐text articles and made copies for RB in which details of the authors and institutions were struck out and the Results section removed: however, despite this RB was able to recognise the studies since this is such a small field in terms of publications. We sought further information from the authors of the primary studies where papers contained insufficient information to make a decision about eligibility.
Data extraction and management
For the 2015 review update, two review authors (JM and MA or CF) independently extracted information using data extraction sheets designed by CF, and resolved discrepancies by discussion. We collected the following information from each study: country where the study was conducted, source of funding, design and methods of the study, study population, inclusion criteria, description of the high‐dose chemotherapy and conventional therapy, outcomes measured and study results. Where possible, we sought missing data from the authors.
Assessment of risk of bias in included studies
Two review authors independently assessed the included studies for risks of bias using the Cochrane 'Risk of bias' assessment tool (www.cochrane‐handbook.org) to assess: selection (random sequence generation and allocation concealment); performance (blinding of participants and personnel); detection (blinding of outcome assessors); attrition (incomplete outcome data); reporting (selective reporting); and other bias. We resolved disagreements by discussion or by recourse to a third review author. We described all judgements fully and presented the conclusions in the 'Risk of bias' table, which was incorporated into the interpretation of review findings by means of sensitivity analyses.
Measures of treatment effect
Some of the available data were immature (e.g. five‐year outcomes were estimated when not all participants had been randomised for five years). Where trialists reported survival rates based on immature data, we used these rates in our tables of comparison. We have noted this in the Characteristics of included studies table of where applicable.
Where results were available only as percentages or only presented in graphs, we calculated the numerators and denominators from the data available and completed the tables of comparison accordingly. We have noted this in the Characteristics of included studies table of where applicable.
Overall survival, event‐free survival, treatment‐related mortality and morbidity are all presented as risk ratios (RRs) with 95% confidence intervals (CIs). We have described quality‐of‐life measures in narrative form.
Several studies have multiple publications and have reported outcomes at different follow‐up times. The data included in this review are the most mature data available for each study.
Unit of analysis issues
All analyses were per woman randomised.
Dealing with missing data
We analysed the data on an intention‐to‐treat basis as far as possible, and tried to obtain missing data from the original trialists. Where these were unobtainable, we did not impute individual values and analysed only the available data.
Assessment of heterogeneity
We assessed statistical heterogeneity by inspecting the scatter in the data points on the graphs and the overlap in their confidence intervals and, more formally, by checking the I² value (Higgins 2003). This measure describes the percentage of total variation across studies that is due to heterogeneity rather than to chance. Interpretation of a given degree of heterogeneity will differ according to whether the estimates show the same direction of effect but we planned to interpret the I² measure as follows: 0% to 40%: might not be important; 30% to 60%: may represent moderate heterogeneity; 50% to 90%: may represent substantial heterogeneity; 75% to 100%: considerable heterogeneity (Higgins 2011).
Assessment of reporting biases
In view of the difficulty of detecting and correcting for publication bias and other reporting biases, we aimed to minimise their potential impact by ensuring a comprehensive search for eligible studies and by being alert for duplication of data. Where there were 10 or more studies in an analysis, we used a funnel plot to explore the possibility of small‐study effects (a tendency for estimates of the intervention effect to be more beneficial in smaller studies).
Data synthesis
We assembled the most complete data set feasible. We assessed clinical heterogeneity between trials through evaluation of potential differences between participants, interventions and outcomes within each study. Where trials appeared to be clinically comparable, we pooled the data to obtain a risk ratio using the Mantel‐Haenszel method in a fixed‐effect model (Mantel 1959), with 95% CIs.
We did not combine quality‐of‐life outcomes because only descriptive data were available.
Subgroup analysis and investigation of heterogeneity
We did not conduct any subgroup analyses. A priori, we had planned to look at the possible contribution of differences in trial design to any meta‐analyses with an I² value of 50% or more.
Sensitivity analysis
We conducted sensitivity analyses by repeating the analyses excluding studies which differed in respect of study quality or chemotherapy regimen, in order to examine the stability of the results.
As there was considerable variation in prognostic factors between participants in the 14 studies, we also conducted a post hoc sensitivity analysis by lymph node status, restricting analysis to studies that included women with 10 or more positive lymph nodes.
Overall quality of the body of evidence: 'Summary of findings' table
We prepared a 'Summary of findings' table using Guideline Development Tool software (GRADEpro GDT). This table highlighted the overall quality of the body of evidence for the main review outcomes, using GRADE criteria (study limitations (i.e. risk of bias), consistency of effect, imprecision, indirectness and publication bias). We justified our judgements about evidence quality (high, moderate or low), and documented and incorporated them into the reporting of results for each outcome.
Results
Description of studies
The included studies differed in several ways, and are described in detail below. Refer to the Characteristics of included studies table.
Results of the search
In the updated search conducted in October 2015, we screened 1385 records, and retrieved 11 potentially relevant articles for further assessment:
-
Nine articles were new publications relating to studies already included in the review (CALGB 2005; Dutch Intergp 2003 x2; GABG 2004; IBCSG 2006; JCOG 2001; MCG 2001; PEGASE 01 2003; WSG 2005).
-
Five of these nine articles reported new data on survival outcomes (IBCSG 2006; GABG 2004; JCOG 2001; MCG 2001) or quality of life (PEGASE 01 2003).
-
The other four articles reported on outcomes not of interest to the current review, and we have added them as additional references to the relevant studies (CALGB 2005; Dutch Intergp 2003 x 2).
-
-
One article was the first publication of a study previously categorised as ongoing (NCT00002772).
-
One study we excluded because it was not an RCT (Sportès 2009).
See PRISMA flowchart (Figure 1).

Study flow diagram.
Altogether we include 14 studies in the updated review and exclude three. All the included studies were randomised trials comparing an experimental group (experimental arm) receiving high‐dose chemotherapy and autologous bone marrow or stem cell transplantation with a control group (control arm) on a lower dose of chemotherapy for the treatment of early poor prognosis breast cancer. All of the included trials have been published in full text.
Included studies
Trial design and setting
Six of the trials were European, three were international, four were North American, and one was Japanese. The 14 trials randomised 5600 women, of whom only 47 (< 1%) were not included in the analyses (see comments on methodological quality below). The five largest trials accounted for 3322 of the women randomised (ACCOG 2004; CALGB 2005; Dutch Intergp 2003; ECOG 2003; NCT00002772). Five of the trials involved between 300 and 400 women (or just over) (IBCSG 2006; GABG 2004; MCG 2001; PEGASE 01 2003; WSG 2005) and one analysed 279 women (ICCG 2005). The other three analysed fewer than 100 women each; in one case this was because it was a pilot study (Dutch pilot 1998) and in another it was due to accrual difficulties (MDACC 2000).
In one study women who relapsed in the control arm became eligible for high‐dose treatment and transplant (CALGB 2005).
Eleven studies reported using stratification, as follows: seven stratified according to menopausal status (CALGB 2005; Dutch Intergp 2003; Dutch pilot 1998; ECOG 2003; IBCSG 2006; ICCG 2005; JCOG 2001), four according to the number of positive axillary nodes (ACCOG 2004; ICCG 2005; JCOG 2001; MCG 2001), six according to participating institution (CALGB 2005; ECOG 2003; IBCSG 2006; ICCG 2005; JCOG 2001, WSG 2005), three according to hormone receptor status (CALGB 2005; ECOG 2003; IBCSG 2006), two according to age (Dutch Intergp 2003; ECOG 2003), two according to clinical stage (CALGB 2005; MDACC 2000), two according to tumour size (Dutch Intergp 2003; WSG 2005), one according to type of primary therapy (NCT00002772) and one according to clinical response to initial chemotherapy (Dutch pilot 1998).
All studies reported their source of funding. Eight studies were funded by non‐profit organisations (CALGB 2005; Dutch pilot 1998; ECOG 2003; GABG 2004; JCOG 2001, MDACC 2000; NCT00002772; PEGASE 01 2003), one by a public health insurance company (Dutch Intergp 2003), one by industry sources (WSG 2005) and four by a combination of non‐profit organisations and industry sources (ACCOG 2004; IBCSG 2006; ICCG 2005; MCG 2001). Four of the studies (CALGB 2005; GABG 2004; IBCSG 2006; MCG 2001) reported that authors had no potential conflict of interest, six (ACCOG 2004; ECOG 2003; MDACC 2000; PEGASE 01 2003; NCT00002772; WSG 2005) reported that one or more of their authors had received some kind of support from pharmaceutical companies, and four studies did not mention whether any of their authors had any potential conflict of interest (Dutch Intergp 2003; Dutch pilot 1998; ICCG 2005; JCOG 2001;).
Participants
All participants were women with early poor prognosis breast cancer, having evidence of multiple axillary lymph node involvement and no evidence of distant metastasis. There was considerable variation between participants in the 14 studies with respect to specific prognostic factors, notably their differing number of positive lymph nodes. Five of the studies included women with four or more positive lymph nodes (ACCOG 2004; Dutch Intergp 2003; ICCG 2005; MCG 2001; PEGASE 01 2003), seven included only women with 10 or more positive nodes (CALGB 2005; ECOG 2003; GABG 2004; JCOG 2001; MDACC 2000; NCT00002772; WSG 2005), one included women with either 10 or more positive nodes or else at least five positive nodes plus an additional risk factor (IBCSG 2006), and the other study (Dutch pilot 1998) specified that women have "extensive axillary node metastasis as evidenced by a positive infraclavicular node biopsy". The number of positive nodes required for inclusion in the trials ranged from "at least four" to "at least ten". In the Dutch Pilot study (1998), a positive axillary apex node on infraclavicular lymph node biopsy was taken as evidence of multiple axillary lymph node involvement.
Median age, where stated, was 43 to 47 years, but ages ranged from 22 to 66 years. The trials used a variety of means to identify and exclude women with distant metastases. Five required participants to have bone marrow aspiration and biopsy (CALGB 2005; ECOG 2003; JCOG 2001; MDACC 2000; NCT00002772); they also required, as did the Dutch Intergp 2003 and the WSG 2005 studies, a normal chest X‐Ray, bone scan and liver ultrasound. The ICCG 2005 study as well as the NCT00002772 study required women to have a normal bone scan. The Dutch pilot 1998 study stated that CT scans and bone marrow biopsies werenot done.
Details of the prognostic characteristics of trial participants are given in Table 1. Table 2 briefly outlines breast cancer staging.
| Study ID | Median Age | Tumour | Median nodes positive | Minimum nodes positive | > 9 nodes | Oestro positive | Progest. positive | Other | Premenop'sal |
| 45 | 3 cm max. | 9 | 4 | 45% | 31% (ER or PR +ve) | 31% (ER or PR +ve) | 43% receptor unknown | ‐ | |
| 45 | 3 cm median | 14 (range 10 ‐ 52) | 10 | 100% | 69% | ‐ | ‐ | ‐ | |
| 45 | T1 5%; T2 30%; T3 45%; T4 10%; Tx 10% | ‐ | N/A: Had pre‐op chemo | N/A | 20% | 25% | 54% receptor unknown | 83% | |
| 45.7 | T1 22%; T2 60%; T3 16% | ‐ | 4 | 35.8% | 65% | 53% | 28% oestrogen receptor negative | ‐ | |
| 44 | ‐ | ‐ | 10 | ‐ | 60% | 59% | 46% > 14 +ve nodes | 72% | |
| ‐ | ‐ | ‐ | 10 | 100% | 60% | 40% | ‐ | 58% | |
| 46 | T1 26%; T2 51%; T3 20% | 13 | 5 ‐ 10 depending on other prognostic factors | 73% | ‐ | ‐ | 40% oestrogen & progesterone receptor ‐ve | 67% | |
| 47 (range 24 ‐ 60) | T1 28%; T2 54%; T3 14%; unknown 4% | 9 (range 4 ‐ 36) | 4 | 45% | 43% | 25% | 38% receptor status not known | 70% | |
| 46 | ‐ | 16 (range 10 ‐ 49) | 10 | 100% | ‐ | ‐ | ‐ | 74% | |
| 45 | ‐ | ‐ | 10 at diagnosis or 4 after initial chemo | > 60% | 50% | 45% | 5% receptor unknown | 68% | |
| ‐ | ‐ | ‐ | 4 | 62% | ‐ | ‐ | ‐ | ‐ | |
| 46 (mean) | ‐ | 13 | 8 | ? | 31% | ‐ | ‐ | 68% | |
| Not stated. 45% were aged 40 ‐ 49 yrs | 20% had T3 tumour | 8% were N2 | ‐ | ‐ | ‐ | ‐ | 66% ER/PgR +ve; 8% receptor unknown | ‐ | |
| 47 | Mean size 3.3 ‐ 3.5 cm | 17 ‐ 18 | 10 | 100% | 63% | ‐ | ‐ | 53% |
+ve = positive
‐ve = negative
ER = estrogen receptor
NA = not applicable
PR = progestogen receptor
| Stage | What stage means |
| I | Breast tumour 2 cm or less in diameter and does not appear to have spread beyond the breast |
| IIA | Breast tumour over 2 cm in diameter OR has spread to the axillary (underarm) lymph nodes on the same side as the breast cancer. The nodes are not stuck to one another or to the surrounding tissues |
| IIB | Breast tumour over 2 cm in diameter AND has spread to the axillary nodes on the same side as the breast cancer. The nodes are not stuck together or to the surrounding tissues. OR the tumour is larger than 5 cm in diameter (and nodes are clear) |
| IIIA | Breast tumour over 5 cms in diameter AND has spread to the axillary lymph nodes on the same side OR tumour has spread to the lymph nodes on the same side as the breast cancer and the nodes are stuck to each other or to the surrounding tissues |
| IIIB | Breast tumour has spread to chest wall or skin OR tumour has spread to internal mammary lymph nodes on the same side as breast tumour |
| IV | Tumour has spread from breast to distant sites or to supraclavicular (above collarbone) lymph nodes |
Most of the studies randomised women soon after full or partial mastectomy and axillary node dissection. The exception was Dutch pilot 1998, which enrolled women for a course of preoperative chemotherapy but excluded before randomisation any women whose disease progressed during the chemotherapy. In one of the American studies (MDACC 2000), women who presented with advanced local disease received a course of preoperative chemotherapy and were eligible for randomisation if they had more than four positive nodes at subsequent surgery. Both Dutch trials, as well as the Japanese trial (JCOG 2001), excluded women who had had prior chemotherapy or radiotherapy. The NCT00002772 trial excluded women who had had radiotherapy, chemotherapy or hormonal therapy for breast cancer or had had chemotherapy for any previous malignancy.
We sought additional information regarding the study design and results from all the principal investigators. We received replies from the Anglo Celtic Oncology Group (ACCOG 2004), the Netherlands Working Party on Autologous Transplantation in Solid Tumours (Dutch Intergp 2003; Dutch pilot 1998), the Eastern Co‐operative Oncology Group (ECOG 2003), the German Breast Cancer Study group (GABG 2004), the MD Anderson group (MDACC 2000), the International Breast Cancer Study Group (IBCSG 2006), the International Collaborative Cancer Group (ICCG 2005), the Michelangelo Cooperative Group (MCG 2001) and the Japanese Clinical Oncology Group (JCOG 2001). However, not all of the requested information was provided: we categorise missing information on trial design as 'Not stated' in the Characteristics of included studies.
Interventions
There was considerable variation among the chemotherapy regimens used. Most of the trials delivered an initial course of chemotherapy at conventional doses to all women. This served as an 'induction' course to women in the high‐dose groups, who went on to receive a high‐dose myeloablative regimen followed by the infusion of stem cells to 'rescue' the bone marrow. In some cases women in the control arms went on to have additional cycles of conventional‐dose chemotherapy and in other cases the conventional treatment was modified in some way to increase the strength or intensity of the dose (CALGB 2005; MCG 2001; WSG 2005). Two trials (IBCSG 2006; MCG 2001) gave the high‐dose arm multiple cycles of high‐dose therapy without a lower‐dose induction course.
-
Initial chemotherapy
In most of the trials both arms received the same initial chemotherapy. Five trials gave all women an initial course of pre‐ and/or postoperative chemotherapy comprising three to four cycles of cyclophosphamide with anthracycline (doxorubicin or epirubicin) and fluorouracil (CALGB 2005; Dutch pilot 1998; Dutch Intergp 2003; ICCG 2005; PEGASE 01 2003). Two trials gave this regimen for six cycles (ECOG 2003; JCOG 2001) and one for eight cycles (MDACC 2000). Two gave multiple cycles of cyclophosphamide and epirubicin only (GABG 2004; WSG 2005), and one (ACCOG 2004) used four cycles of doxorubicin alone for the initial chemotherapy. Doses in the control arm varied: see Table 3. During this initial phase of chemotherapy, women randomised to receive the high‐dose treatment had a course of granulocyte colony‐stimulating factor (GCSF) to stimulate the production of white cells which were then harvested for later transplantation.
| Study | Phase 1 | Phase 2 |
| doxorubicin 75 mg | cyclophosphamide | |
| cyclophosphamide 600 mg | cyclophosphamide 900 mg | |
| cyclophosphamide 500 mg | ‐ | |
| cyclophosphamide 500 mg | ‐ | |
| cyclophosphamide 1400 mg (po) | ‐ | |
| cyclophosphamide 600 mg | cyclophosphamide 1 gm | |
| doxorubicin 60mg or epirubicin 90 mg | cyclophosphamide 1400 mg (po) | |
| cyclophosphamide 600 mg | cyclophosphamide 1200 mg | |
| cyclophosphamide 500 mg | ‐ | |
| epirubicin 120 mg 3 cycles | cyclophosphamide 600 mg | |
| cyclophosphamide 500 mg | ‐ | |
| cyclophosphamide 500 mg | ‐ | |
| sequential administration of 3 cycles each of doxorubicin 80 mg/m², paclitaxel 200 mg/m², and cyclophosphamide | ‐ | |
| cyclophosphamide 600 mg | cyclophosphamide 600 mg |
-
The control arm
In five of the trials the women in the control arm had no further chemotherapy after the initial phase mentioned above. In two (Dutch Intergp 2003; ICCG 2005) the control arm had a continuation of the initial chemotherapy. In two trials (ACCOG 2004; GABG 2004) they received a standard course of a different chemotherapy, cyclophosphamide, methotrexate and fluorouracil (CMF). The control arm in one trial (CALGB 2005) were given an "intermediate level" version of the high‐dose therapy along with a course of GCSF to stimulate white cell production. In another (WSG 2005) they had a dose‐dense regimen supported by GCSF, comprising two further cycles of the initial chemotherapy followed by three cycles of CMF, all at two‐week intervals.
In three trials where the two arms did not have any of their treatment in common, the control arm received two combination therapies in sequence: in the Italian trial (MCG 2001) the control arm received three cycles of epirubicin followed by six of CMF; in the IBCSG 2006 trial they received four cycles of epirubicin and cyclophosphamide (EC) followed by three cycles of CMF. In NCT00002772 the control group received sequential dose‐dense and dose‐escalated chemotherapy consisting of three cycles of doxorubicin, paclitaxel, and cyclophosphamide, with filgrastim support. The study authors noted that this regimen is non‐standard.
-
The experimental arm
In most studies, after the initial chemotherapy described above, the experimental arm went on to receive one or two cycles of high‐dose chemotherapy. The high‐dose therapy for most trials comprised cyclophosphamide with thiotepa or etoposide or carmustine, with or without a platinum‐based drug (cisplatin or carboplatin) or mitoxantrone. Doses varied: see Table 4 and Characteristics of included studies tables.
| Study | Initial phase | High‐dose cycle 1 | High‐dose cycle 2 | High‐dose cycle 3 | High‐dose cycle 4 | Regimen |
| 4 cycles of doxorubicin (as control arm) followed by: | cyclophosphamide 4 gm | cyclophosphamide 6 gm | ‐ | ‐ | Divided doses over 4 days | |
| 4 cycles of cyclophosphamide, doxorubicin and fluorouracil (as control arm) followed by: | cyclophosphamide 5.625 gm | ‐ | ‐ | ‐ | Divided doses over 3 days | |
| 4 cycles of cyclophosphamide, epirubicin and fluorouracil (doses as control arm) followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Divided doses over 4 days | |
| 4 cycles of cyclophosphamide, epirubicin and fluorouracil (as control arm) followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Divided doses over 4 days | |
| 6 cycles of cyclophosphamide, doxorubicin and 5FU (as control arm) followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Continuous infusion over 4 days | |
| 4 cycles of cyclophosphamide and epirubicin (as control arm) followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Divided doses over 4 days | |
| No common path with control group protocol | epirubicin 200 mg | As cycle 1 | As cycle 1 | 3 X 21‐day cycles | ||
| 2 cycles of cyclophosphamide, epirubicin and fluorouracil (as control arm cycles 1 and 2) | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Continuous infusion over 4 days | |
| 6 cycles of cyclophosphamide, doxorubicin and fluorouracil (as control arm), followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | ‐ | |
| No common path with control group protocol | cyclophosphamide 7 gm | methotrexate 8gm | epirubicin 120 mg X 2 | thiotepa 600 mg melphalan 160 ‐ 180 mg | 4 high‐dose treatments in sequence | |
| 8 cycles of cyclophosphamide, doxorubicin and fluorouracil (as control arm), followed by: | cyclophosphamide 5.25 gm | As cycle 1 | ‐ | ‐ | Divided doses over 3 days. 2nd cycle given when haematologically safe | |
| 4 cycles of cyclophosphamide, epirubicin and fluorouracil (as control arm), followed by: | cyclophosphamide 120 mg | ‐ | ‐ | ‐ | ‐ | |
| 4 cycles of doxorubicin 80 mg/m² and cyclophosphamide 600 mg/m² (AC) every 3 weeks | STAMP I or STAMP V HDC regimen. STAMP I consisted of cyclophosphamide 1.85 g/m²/d and cisplatin 55 mg/m²/d, followed by carmustine 600 mg/m²; STAMP V consisted of cyclophosphamide 1.5 g/m²/d, carboplatin 200 mg/m²/d, and thiotepa 125 mg/m²/d | ‐ | ‐ | ‐ | ‐ | |
| 2 cycles of cyclophosphamide and epirubicin (as control arm) | cyclophosphamide 3 gm | As cycle 1 | ‐ | ‐ | High‐dose cycles over 28 days |
As noted above, three studies differed in design by giving different initial chemotherapy to the experimental arm. In one (MCG 2001) the experimental arm had a sequence of cyclophosphamide, methotrexate, epirubicin and thiotepa with melphalan, all at high doses, and in the second (IBCSG 2006) they had three cycles of high‐dose epirubicin and cyclophosphamide; in the third (NCT00002772) they received four cycles of doxorubicin and cyclophosphamide.
In all cases high‐dose therapy was supported by autologous peripheral blood progenitor cell transplantation or bone marrow transplantation or both, using the cells or marrow harvested during the initial phase of chemotherapy.
-
Radiotherapy
All women in 10 of the trials received a course of loco‐regional radiotherapy after chemotherapy (ACCOG 2004; CALGB 2005; Dutch Intergp 2003; Dutch pilot 1998; ECOG 2003; ICCG 2005; MDACC 2000; PEGASE 01 2003; NCT00002772; WSG 2005), and this was introduced as a protocol change in another study (GABG 2004). In the Italian trial (MCG 2001) only women who had had conservative surgery received radiotherapy, and in another (IBCSG 2006) it was mandatory after breast‐conserving surgery but recommended for all women. The Japanese trial (JCOG 2001) did not include radiotherapy as part of the protocol.
-
Tamoxifen
Trial protocols for tamoxifen varied. It was prescribed for all women in seven trials (ACCOG 2004; Dutch Intergp 2003; Dutch pilot 1998; IBCSG 2006; ICCG 2005; JCOG 2001; MCG 2001). Other trials did not prescribe it for women who had hormone receptor‐negative disease (CALGB 2005; ECOG 2003; GABG 2004; WSG 2005) or were premenopausal (MDACC 2000; PEGASE 01 2003; NCT00002772). The duration of treatment, where specified, was five years for all trials except Dutch Intergp 2003 (in which the duration of treatment was initially two years but was changed to five years during the course of the trial), Dutch pilot 1998 (in which the duration of treatment was two years) and JCOG 2001 (in which the duration of treatment was "at least two years").
Outcomes
All studies measured overall survival (i.e. survival with or without recurrence), and all specified the number of deaths caused by treatment toxicity.
All studies measured event‐free survival (i.e. survival without breast cancer recurrence). Two studies differed by including the incidence of other cancers in events for this outcome, without separately reporting the data relating to breast cancer alone (CALGB 2005; GABG 2004).
With regard to non‐fatal toxicity, eight studies (Dutch Intergp 2003; Dutch pilot 1998; ACCOG 2004; ECOG 2003; ICCG 2005; MDACC 2000; NCT00002772; WSG 2005) described the side effects experienced by women on both standard and high‐dose regimens. Six trials reported long‐term toxicity which included the incidence of second cancers (CALGB 2005; Dutch Intergp 2003; Dutch pilot 1998; IBCSG 2006; ICCG 2005; MDACC 2000).
Limited quality‐of‐life data have been published (ACCOG 2004; CALGB 2005; Dutch Intergp 2003; Dutch pilot 1998; PEGASE 01 2003; WSG 2005).
Mature data on overall survival and event‐free survival have been published for five studies. Four of these have followed up all women for three years (Dutch Intergp 2003; MDACC 2000; PEGASE 01 2003; WSG 2005) and one has followed up all women for five years (Dutch pilot 1998). The other studies had median follow‐up times ranging from three to over 11 years and reported estimated survival rates at differing follow‐up periods, based on their results to date. See Table 5.
| Study ID | Data maturity | Median follow‐up |
| No | 4 years | |
| No | 7.3 years | |
| 5 years | 6.9 years | |
| 3 years | 7 years | |
| No | 6.1 years | |
| No | 6.1 years | |
| No | 8.3 years | |
| No | 4.2 years | |
| No | 63 months | |
| 3 years | 11.9 years | |
| No | 11.33 years | |
| 3 years | 3.25 years | |
| No | 5.8 years | |
| 3 years | 4 years |
Only six studies (Dutch Intergp 2003; Dutch pilot 1998; ECOG 2003; ICCG 2005; MDACC 2000; NCT00002772) reported data comparing the adverse effects of the different doses after the end of chemotherapy or during long‐term follow‐up, or both.
Excluded studies
We excluded three studies, one because it compared two experimental chemotherapy regimens and did not include a control group receiving conventional chemotherapy (Bergh 2000), one because the trial results have been retracted after an investigation for breach of scientific integrity (Bezwoda 1999), and one because it was not an RCT (Sportès 2009).
Risk of bias in included studies
Allocation
Sequence generation
We rated 10 trials at low risk of bias for sequence generation; all used computerised methods. The other four trials did not describe the methods used, and we rated them at unclear risk.
Allocation concealment
We rated 10 trials at low risk of bias for allocation concealment; most used remote allocation. The other four trials did not describe the methods used, and we rated them at unclear risk.
Blinding
Dutch Intergp 2003 conducted a centralised review of pathological specimens in a blinded fashion, but otherwise blinding was not mentioned in any group. As it appears unlikely (but not impossible) that blinding would influence our primary review outcomes, we rated all studies at unclear risk of bias in this domain.
Incomplete outcome data
We rated all studies at low risk of bias in this domain, as in all cases 95% to 100% of women randomised were included in the analysis.
Selective reporting
All studies reported all expected outcomes and were rated at low risk of bias in this domain.
Other potential sources of bias
We identified no other potential source of bias for 10 studies, and we rated them at low risk for this domain. Four studies reported issues that could potentially cause bias, and we rated these at unclear risk.
For details on risks of bias please see Figure 2; Figure 3 and Characteristics of included studies

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
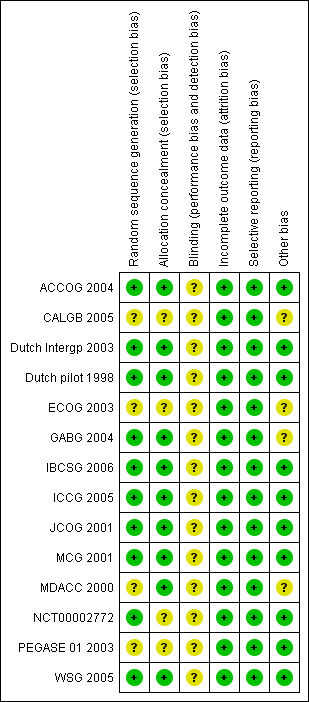
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
A total of 5600 women were analysed in 14 studies, of whom 2800 were randomised to receive high‐dose chemotherapy with stem cell transplantation (experimental group) and 2800 were randomised to conventional treatment (control group).
High‐dose chemotherapy with autograft versus chemotherapy without bone marrow transplant or stem cell rescue
Primary outcomes
1. Overall survival
Refer to Figure 4.
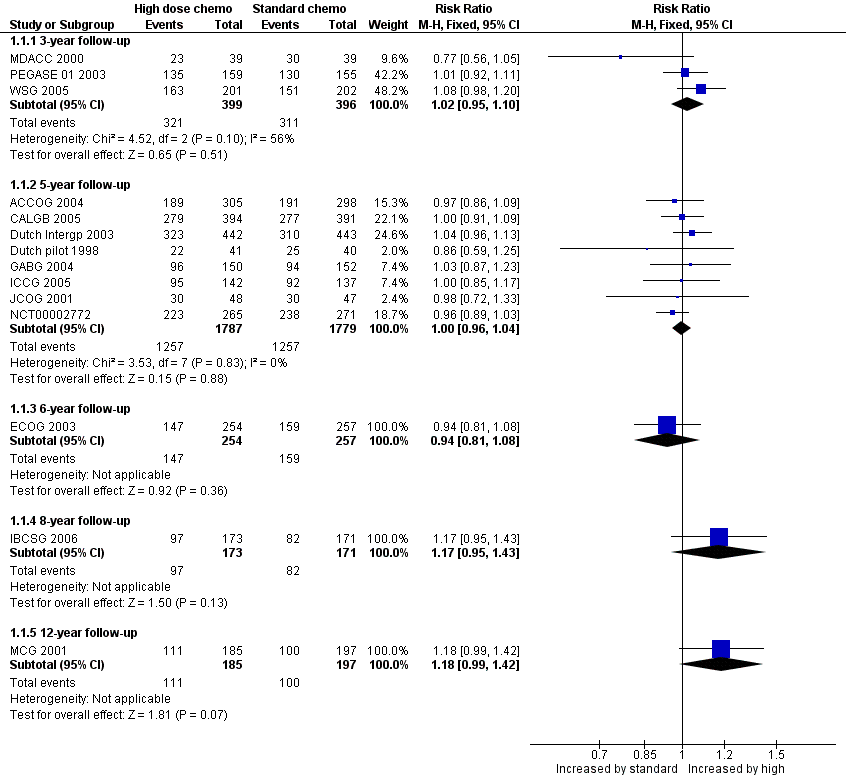
Forest plot of comparison: 1 High‐dose chemotherapy versus standard chemotherapy, outcome: 1.1 Overall survival.
Three‐year follow‐up
We pooled three studies for this outcome. There was no evidence of a difference between the groups (risk ratio (RR) 1.02, 95% confidence interval (CI) 0.95 to 1.10, 3 RCTs, 795 women, I² = 56%; Analysis 1.1). There was moderate statistical heterogeneity for this finding. Heterogeneity appeared to be attributable to MDACC 2000, which was the smallest study in the review but did not differ from other studies in any obvious way. Exclusion of this study from analysis did not substantially affect the findings (RR 1.05, 95% CI 0.98 to 1.13, 2 RCTs, 717 women, I² = 0%).
Five‐year follow‐up
We pooled eight studies for this outcome. There was no evidence of a difference between the groups (RR 1.00, 95% CI 0.96 to 1.04, 8 RCTs, 3566 women; I² = 0%; Analysis 1.1).
Six‐year follow‐up
One study (ECOG 2003) reported this outcome. There was no evidence of a difference between the groups (RR 0.94, 95% CI 0.81 to 1.08, 1 RCT, 511 women; Analysis 1.1).
Eight‐year follow‐up
One study (IBCSG 2006) reported this outcome. There was no evidence of a difference between the groups (RR 1.17, 95% CI 0.95 to 1.43, 1 RCT, 344 women; Analysis 1.1).
12‐year follow‐up
One study (MCG 2001) reported this outcome.There was no evidence of a difference between the groups (RR 1.18, 95% CI 0.99 to 1.42, 1 RCT, 382 women; Analysis 1.1).
2. Event‐free survival
Refer to Figure 5.

Forest plot of comparison: 1 High‐dose chemotherapy versus standard chemotherapy, outcome: 1.2 Event‐free survival.
Three‐year follow‐up
We pooled three studies for this outcome. There was a statistically significant difference between the groups, favouring the high‐dose group (RR 1.19, 95% CI 1.06 to 1.34, 3 RCTs, 795 participants, I² = 56%; Analysis 1.2). There was moderate statistical heterogeneity for this finding. Heterogeneity appeared to be attributable to MDACC 2000, which was the smallest study in the review but did not differ from other studies in any obvious way. Exclusion of this study from analysis did not substantially affect the findings (RR 1.24, 95% CI 1.10 to 1.40, 2 RCTs, 717 women, I² = 0%).
Five‐year follow‐up
We pooled eight studies for this outcome. There was no evidence of a difference between the groups (RR 1.04, 95% CI 0.99 to 1.10, 9 RCTs, 3566 women, I² = 25%; Analysis 1.2).
Six‐year follow‐up
One study (ECOG 2003) reported this outcome. There was no evidence of a difference between the groups (RR 1.04 (95% CI 0.87 to 1.24, 1 RCT, 511 women; Analysis 1.2).
Eight‐year follow‐up
One study (IBCSG 2006) reported this outcome. There was no evidence of a difference between the groups (RR 1.27, 95% CI 0.99 to 1.64, 1 RCT, 344 women; Analysis 1.2).
12‐year follow‐up
One study (MCG 2001) reported this outcome.There was no evidence of a difference between the groups (RR 1.18, 95% CI 0.95 to 1.45, 1 RCT, 382 women; Analysis 1.2).
Secondary outcomes
1. Treatment‐related deaths
There were 68 deaths attributed to treatment toxicity among the 2800 women who were randomised to receive high‐dose chemotherapy with stem cell transplantation, and five among the 2800 women in the control arms. There were significantly fewer treatment‐related deaths in the control group (RR 7.97, 95% CI 3.99 to 15.92, 14 RCTs, 5600 women, I² = 12%; Analysis 1.3). There were no treatment‐related deaths in three of the trials (Dutch pilot 1998; JCOG 2001; WSG 2005).
Treatment‐related deaths were accounted for as follows:
-
Deaths in the high‐dose arm: most of the deaths occurred in one study (CALGB 2005), where there were 33 treatment‐related deaths in the high‐dose arm (33/394). Most of the deaths in this study were caused by acute infection, pulmonary toxicity or renal failure (haemolytic‐uraemic syndrome). However there were also three late treatment‐related deaths, one due to acute myeloid leukaemia secondary to treatment and two due to pulmonary fibrosis. There were five treatment‐related deaths in the high‐dose arm in the ACCOG 2004 study, four related to infection and one to pulmonary fibrosis (5/307). In the Dutch Intergp 2003 trial one woman died of cardiac arrhythmia during the preliminary standard‐dose chemotherapy (before receiving high‐dose treatment) and a further four women died within 100 days of autograft, two of septicaemia and two from cardiac problems (5/442). Nine women died in the ECOG 2003 study within eight weeks of stem cell transplantation; in six cases these women had been given stem cells from the bone marrow rather than from the peripheral circulation (9/254). There was one death during the transplant procedure in the PEGASE 01 2003 trial, (1/159) and one death from interstitial pneumonia in the Italian trial (MCG 2001) (1/185). In the small American trial (MDACC 2000) one woman died from treatment‐related sepsis (1/39). There were two acute and two late treatment‐related deaths in the IBCSG 2006 trial (4/173). Two women in the ICCG 2005 trial died of liver failure caused by hepatic veno‐occlusive disease and one died from cardiomyopathy (3/142). In the German trial (GABG 2004) there were three treatment‐related deaths, caused by lung toxicity, cardiac toxicity and acute myeloid leukaemia (suspected to be chemotherapy‐induced) (3/150). Three deaths occurred as a result of treatment‐related toxicity in NCT00002772, one due to acute respiratory distress syndrome during induction of chemotherapy and two during transplantation (one from pneumonia and the other from complications of veno‐occlusive disease).
-
Deaths in the standard‐dose arm: Five women in the standard‐dose groups died as a result of treatment toxicity, two in the ICCG 2005 trial (2/137), two in the GABG 2004 trial (2/152), and one in the NCT00002772 trial, in which the cause of death was a cardiac event (1/271). In the ICCG 2005 trial one woman died three months after randomisation as a result of neutropenia associated with colitis, and another died suddenly 10 days after chemotherapy, possibly due to anthracycline cardiotoxicity.
2. Morbidity
Five studies described the adverse effects of conventional‐dose chemotherapy (which was given to all women in these studies), and also described the additional toxicity experienced by women who went on to receive high‐dose therapy (Dutch Intergp 2003; Dutch pilot 1998; ECOG 2003; ICCG 2005; MDACC 2000). Three studies compared toxicity between the control and high‐dose arms (ACCOG 2004; NCT00002772; WSG 2005). Another study, in which the women in the two arms had no treatment in common (IBCSG 2006), compared the worst toxic effects experienced by the two arms. The German study (GABG 2004) systematically registered toxicity only in the high‐dose arm.
Toxicity was much higher in the high‐dose group, notably (as expected) neutropenia, often accompanied by fever and infection. The Dutch pilot 1998 study was the only one to report on the length of the hospital inpatient stay for high‐dose therapy: 90% of women required up to 18 days in hospital after transplantation to allow their bone marrow to regenerate.
Classic treatment side‐effects such as fatigue, vomiting, mucositis and diarrhoea were common with both regimens, although all were more common and more severe with high‐dose treatment. Two studies mentioned hair loss, which was universal in the high‐dose groups. High‐dose therapy was also more likely to induce menopause. Three studies mentioned that about 75% to 80% of women in the high‐dose arm and 60% of women in the control arm were postmenopausal after therapy (Dutch Intergp 2003; IBCSG 2006; PEGASE 01 2003), while another noted that all women in the high‐dose arm became amenorrhoeic (WSG 2005). All premenopausal participants in ICCG 2005 became amenorrhoeic after chemotherapy.
Organ toxicities affected more women in the high‐dose arms; these included cardiac, pulmonary, renal, hepatic, bladder, skin and neurological complications. Most of these toxicities were not severe and were reversible; one study reported no major organ toxicities in either arm (WSG 2005). However, some toxicities were fatal, as described above, and in addition there were a number of women who suffered ongoing treatment‐related morbidities such as peripheral neuropathy, congestive heart failure, pulmonary fibrosis and radiation‐induced pneumonitis; such long‐term complications affected women in both groups but were more common in the high‐dose group. See Table 6 for details.
| Study ID | Haemopoietic | Gastrointestinal | Pulmonary | Cardiac events | Neurological | Other toxicity | Late/ long term | Second cancers | Trialist's summary |
| Standard chemo: Grade 4 neutropenia 15% | Haemorrhage ≥ grade 2: | Nausea ≥ grade 3: | Rhythm toxicity ≥ grade 2: | Cortical neurotoxicity ≥ 1 | Both trial arms: Menopausal symptoms common. | ‐ | ‐ | ‐ | |
| Leukopenia and thrombocytopenia common in both groups but more severe and persistent in HDC arm | ‐ | Toxicity ≥ grade 3: | ‐ | Toxicity ≥ grade 3: | Hepatic toxicity ≥ grade 3: | ‐ | By median 7.5 yrs High‐dose arm: 16 second cancers (4%) (including acute myeloid leukaemia or myelodysplatic syndrome 7; breast cancer 5) | ‐ | |
| High‐dose chemo: all hospitalised for 13 ‐ 30 days for haemopoietic recovery. Median neutropenic fever 5 days Standard chemo: neutropenic fever after 4% of cycles | High‐dose: mucositis 85% (severe in 22%), diarrhoea common. Standard chemo: Mild nausea and vomiting, mucositis (28% of cycles), diarrhoea (4% of cycles) | ‐ | See long‐term events | ‐ | Both arms: alopecia 100%, fatigue common, lymphoedema of arm in 20% High‐dose: ovarian failure 100%, radiation pneumonitis 10%, Standard dose: radiation pneumonitis 2% | High‐dose arm: 1 case hypothyroidism, 1 case auto‐antibody production | At median follow‐up of 7 years: | High‐dose: "Moderately well tolerated but substantial though reversible toxic effects". Standard dose: "Mild toxicity" | |
| High‐dose: transfusion‐dependent 100% | High‐dose: nausea and vomiting 100% | ‐ | High‐dose: cardiac arrhythmia 1/442, possible heart failure 1/442 | ‐ | High‐dose: high fever (necessitating early termination of treatment): 4 women (1%) | ‐ | By median follow‐up 7 years: | High‐dose: "Well tolerated" | |
| High‐dose: leukopenia 98%, granulocytopenia 94%, thrombocytopenia 97%, anaemia 62%, | High‐dose: nausea 32%, vomiting 16%, diarrhoea 22%, stomatitis 37% Standard chemo: nausea 11%, vomiting 8%, stomatitis 4% (all grade 3 or 4) | Standard chemo: 1% (grade 3 or 4) | ‐ | Standard chemo: 6% (grade 3 or 4) | High‐dose: infection 21%, liver effects 13%, skin effects 11%, diabetes 14% Standard dose: hyperglycaemia 2%, phlebitis 1%, hepatotoxicity 1% (all grade 3 or 4) | ‐ | By median 6.1 years: | ‐ | |
| ‐ | High‐dose: Grade 3 or 4 gastrointestinal toxicity < 1%; | Grade 3 or 4 toxicity < 1% | ‐ | High‐dose: Grade 3 or 4 toxicity nil | High‐dose: Grade 3 or 4 toxicity: Bladder < 1%; kidney nil; liver nil | ‐ | ‐ | ‐ | |
| High‐dose: myelosuppression | High‐dose: nausea and vomiting; mucositis | ‐ | ‐ | ‐ | ‐ | Permanent amenorrhoea: High‐dose arm 77/95 (81% overall, age < 40 years 61%; age > 40 years 96%); Standard‐dose arm 61/98 (63% overall age < 40 years 24%; age > 40 years 84%) | By median 8.3 years: | High‐dose: Overall toxicities Grade 3 1%; Grade 4 98%; | |
| High‐dose: leucopenia and thrombocytopenia presumed 100% but nadir count not always available (grade 3 or 4) | High‐dose: nausea and vomiting 46%, mucositis 22% (grade 3 or 4) | High‐dose: Pulmonary embolus 1/143; respiratory failure requiring ventilator 1/143 | High‐dose: severe cardiac arrhythmia 2% (3/143) | ‐ | High‐dose: hair loss 100%, fever (no infection) 17%, infection 24%, "other" 28% (grade 3 or 4), deep vein thrombosis 1/143 | After chemotherapy: 227 toxic events occurred (127 in high‐dose arm, 110 in control arm), of which 30% related to tamoxifen. Of the others, 7 events deemed life‐threatening (5 in high‐dose group, 2 in control arm) | High‐dose: 2/143 (breast 1, ovarian 1) | ||
| High‐dose: All 34 women receiving HDC actually developed grade 4 leukopenia and grade 4 neutropenia; 27 (79%) developed grade 4 and the other 7 grade 3 thrombocytopenia. Standard dose: 7 women (8%) developed grade 4 neutropenia | High‐dose: vomiting 62%, diarrhoea 29%, mucositis 15%, (grade 3 or 4) | ‐ | High‐dose: grade 3 arrhythmia 3%, | ‐ | High‐dose: Grade 3 or 4 infection: 6% | ‐ | ‐ | ‐ | |
| High‐dose: Length of hospital stay not stated. Standard dose: 22% admitted with infection or fever | High‐dose: mild/moderate vomiting 80%, mild/moderate diarrhoea 58%, mild/moderate mucositis 83%. Standard dose: Nausea and vomiting moderate 75%, severe 16%. Diarrhoea moderate 19%, severe 8%. Mucousitis moderate 36%, severe 10% | High‐dose: 1 case (severe) | High‐dose: moderate/severe 8%. Standard dose: 1 woman (1%) had myocardial infarction | High‐dose: hearing loss 2 cases (6%) ‐ 1 permanent, mild/moderate peripheral neuropathy 11% | High‐dose: Renal: 25% (22% mild, < 3% severe), hepatic (mild/moderate) 31%, bladder (moderate) 25%, skin (mild) 8% | High‐dose: 1 case of avascular necrosis | High‐dose: 1 case of acute myeloid leukaemia | "Overall there was greater and more frequent morbidity associated with high dose chemotherapy" | |
| ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| High‐dose: 62% had haematologic toxicity during induction and 92% had it during transplantation. 3 women had myelodysplastic syndrome Controls: 59% had haematologic toxicity 2 women had myelodysplastic syndrome | ‐ | ‐ | ‐ | ‐ | ‐ | High‐dose: 44% of women experienced grade 3 or 4 nonhaematologic toxicity during induction while 80% experienced grade 3 or 4 nonhematologic toxicity during transplantation. Control arm: Approximately 63% experienced grade 3 or 4 nonhaematologic toxicity, most commonly fatigue, nausea and vomiting, infection, febrile neutropenia, mucositis, and sensory neuropathy | ‐ | High‐dose: 44% had | |
| ‐ | High‐dose arm: nausea 25%, mucositis 18%, diarrhoea 5% | High‐dose arm: 1% | High‐dose arm: 3% | ‐ | High‐dose arm: grade 3 or 4 skin toxicity 3%, amenorrhoea 100% | ‐ | ‐ | Both high‐dose chemotherapy and dose‐dense conventional chemotherapy are feasible with tolerable toxicity in a multicentre setting |
Several trials compared the incidence of second primary malignancies in women from the two trial arms at variable durations of follow‐up (CALGB 2005; Dutch Intergp 2003; Dutch pilot 1998; IBCSG 2006, ICCG 2005; MDACC 2000; NCT00002772). These included second breast cancers and haematological malignancies as well as other types of cancer. A meta‐analysis of these results showed no significant difference between the two arms in the incidence of second cancers at a median of four to nine years of follow‐up (RR 1.25, 95% CI 0.90 to 1.73, 7 RCTs, 3423 women, I² = 0%; Analysis 1.4).
3. Quality of Life
Women from five of the trials took part in related quality‐of‐life studies (ACCOG 2004; CALGB 2005; Dutch Intergp 2003; PEGASE 01 2003, WSG 2005) and in addition a subset of women drawn from both Dutch trials participated in a study of chemotherapy‐related cognitive impairment.
The ACCOG 2004 study analysed quality‐of‐life data relating to 84 women on the high‐dose treatment arm and 82 on the standard‐dose arm. There was no significant difference between the groups: at six months women in both arms of the trial reported a significant deterioration in quality of life, but at one year both groups reported a quality of life very similar to the pre‐treatment baseline. Both groups were significantly less tense and worried at follow‐up assessments than they were at baseline. At five years the investigators reported that quality of life associated with high‐dose therapy was only transiently lower than that associated with conventional dose therapy. More detailed analysis is planned.
A subset of 210 women from the CALGB 2005 trial was enrolled in a companion quality‐of‐life study, using telephone‐based questionnaires. The high‐dose group reported significantly worse overall quality of life at three months, but the difference between the groups narrowed until there was minimal difference among survivors at one year. Quality of life improved in both groups over the follow‐up time.
Quality of life data were also available from 96 women in the WSG 2005 trial. Again, quality of life scores were worse in the high‐dose arm but recovered by three weeks after the second cycle of high‐dose therapy. This outcome was not followed up beyond that point.
Women participating in the Dutch Intergp 2003 trial were sent regular quality‐of‐life questionnaires which were completed for at least four years by 58 women in the conventional arm and 46 in the high‐dose arm. The high‐dose arm scored significantly lower on several measures just after chemotherapy, but there was no significant difference between the arms six months later. Scores improved consistently in both arms over time with no significant difference between them. However, at four years more than 20% of women in both groups reported fatigue, sore muscles, decreased sexual interest and sweating as adverse effects of therapy.
A further study in the Dutch Intergp 2003 trial surveyed 413 women three years after chemotherapy. There was no difference between the two arms in their mean vitality score at one, two or three years, and their scores never fell below those of the reference population. Women in the high‐dose arm had a slightly lower haemoglobin level during the three years following chemotherapy, but overall this did not correlate well with reports of fatigue: 81% of women reporting fatigue did not have a low haemoglobin.
The PEGASE 01 2003 trialists found that women in the high‐dose arm recorded a strong deterioration in quality of life during treatment and that physical functioning was significantly better for women in the control arm. At three months after completion of radiotherapy the differences between the two arms had disappeared. However, at one year follow‐up women in the high‐dose arm had physical and functional scores below their baseline values, which were significantly lower than the scores of women in the control arm.
The effect of chemotherapy on cognitive functioning was evaluated in a report in which 102 women took part, 34 from the high‐dose arm of either one of the Dutch trials, 34 from the standard‐dose arm of the same trials, and 34 from a control group of women with stage I breast cancer not treated with chemotherapy. Psychoneurological tests administered at a median of two years after treatment showed cognitive impairment in 32% of the high‐dose group, 17% of the standard‐dose group and 9% of the control group. The difference between the high‐dose group and the control group was statistically significant (P = 0.006) but the difference between the high‐dose arm and the standard‐dose group was not (P = 0.056). It was observed that the women who reported cognitive problems were not necessarily the same ones who were objectively identified as being cognitively impaired, and that self‐reported cognitive problems were more related to anxiety, depression and psychological distress. At four years the tests were repeated with a subset of the original participants and the results suggested that cognitive dysfunction after chemotherapy may be transient; however, there was a high attrition rate of initially cognitively impaired women in the high‐dose arm.
Statistical heterogeneity
We noted moderate or high statistical heterogeneity in two instances: namely, event‐free survival and overall survival at three years. In both cases the results of MDACC 2000 differed in direction from those of other trials, although the 95% confidence interval overlapped the confidence interval of the summary effect measure. When MDACC 2000 was omitted from the meta‐analyses, there was no statistically significant heterogeneity. It is unclear why the results of MDACC 2000 were relatively less favourable for the high‐dose arm; however, it was the smallest trial in the review, which limits the strength of its findings.
Sensitivity analyses
-
Study quality:
We conducted sensitivity analyses excluding studies which did not clearly report satisfactory methods of randomisation and allocation concealment (CALGB 2005; ECOG 2003; MDACC 2000; PEGASE 01 2003), studies which did not state that they analysed all randomised participants by intention‐to‐treat (MCG 2001) and studies which did not indicate that prognostic factors were balanced between the two arms (MCG 2001; JCOG 2001). These analyses did not change the statistical significance of the results.
-
Chemotherapy regimen:
Over half of the treatment‐related deaths were in CALGB 2005, which noted that deaths occurred more frequently in centres that did fewer than 50 transplants. It was one of only two studies to use carmustine as part of the high‐dose regimen. It differed from the other studies in that women in the conventional arm of the study were given their final cycle of chemotherapy at an "intermediate" dose with GCSF (growth factor) support, and has been described as "a comparison between high and intermediate dose" chemotherapy regimens (Antman 1992). Excluding CALGB 2005 from the analysis did not change the statistical significance of any of the results.
As noted above, there was considerable variation between the chemotherapy regimens used. We conducted sensitivity analyses omitting firstly studies that used upfront high‐dose chemotherapy with no induction cycles for the high dose group (IBCSG 2006; MCG 2001), and secondly studies where the 'conventional' chemotherapy was modified in some way to increase the strength or intensity of the dose (CALGB 2005;MCG 2001;NCT00002772WSG 2005). In neither case did these omissions affect the statistical significance of the results.
-
Lymph node status
Sensitivity analysis restricted to studies that randomised women with 10 or more nodes (with and without the Dutch pilot 1998 study and IBCSG 2006 in both groups) did not affect the statistical significance of the results.
Assessment of publication bias
A funnel plot for the outcome of treatment‐related mortality was not suggestive of publication bias (Figure 6).

Funnel plot of comparison: 1 High‐dose chemotherapy versus standard chemotherapy, outcome: 1.3 Treatment‐related mortality.
Discussion
Summary of main results
These studies tested the hypothesis that women with early poor prognosis breast cancer would benefit from treatment with high‐dose chemotherapy with autologous bone marrow or stem cell support. There was statistically significant evidence of increased event‐free survival for women in the high‐dose group at three year follow‐up, but there was no good evidence of a difference between the groups at other time points. There was no evidence of a difference between the groups in overall survival rates at any stage of follow‐up and in fact there was evidence of harm, with greater numbers of treatment‐related deaths and adverse events occurring in the high‐dose arms.
CALGB 2005 dominates the results with respect to treatment‐related deaths: the increased mortality is thought to have occurred because of the pulmonary and hepatic toxicity of carmustine used in this study and the relative inexperience of some transplant centres. However, sensitivity analysis excluding this trial did not negate the statistical significance of the results for this outcome, which continued to favour the group that received conventional chemotherapy.
Overall completeness and applicability of evidence
Many researchers have suggested that while most studies have not demonstrated an overall benefit, there may be subgroups that benefit from high‐dose therapy. The Dutch Intergp 2003 study reported improved event‐free survival at five years in women with more than 10 positive nodes in the high‐dose arm that was of borderline statistical significance by the log‐rank test (P = 0.05, hazard ratio for relapse 0.71 (95% CI 0.5 to 1.0)). However, two of the other studies in this review reported survival rates for women with at least 10 nodes at five and six years respectively, and neither showed any statistically significant advantage for the high‐dose group (CALGB 2005; ECOG 2003). Nor did subgroup analyses of women with 10 or more positive lymph nodes in two other trials (ACCOG 2004; IBCSG 2006) find any statistically significant survival benefit for the high‐dose group.
Unplanned subgroup analyses in the Dutch Intergp 2003 study unexpectedly showed a statistically significant benefit in event‐free survival for women in the high‐dose group with lower expression of the HER2/neu gene, which is associated with cell division (P = 0.002 at five‐year follow‐up). Younger age and lower histological grade were also associated with more responsiveness to high‐dose chemotherapy in this study. Retrospective analysis of data from the German study (WSG 2005) found that younger women with large higher‐grade tumours benefited most from high‐dose treatment in their study, although subgroups were very small. More recently, this group has proposed a specific biomarker (Y‐box binding protein YB‐1) as a method of identifying women who might benefit from high‐dose chemotherapy (Gluz 2009). As the authors noted, their subgroup analyses were unplanned (and therefore must be viewed with caution), but may indicate promising areas for investigation. No evidence of a difference between the groups was reported in other subgroup analyses of baseline prognostic factors such as age, tumour size and grade, menopausal status, surgery type and hormone receptor status (ACCOG 2004; ECOG 2003; IBCSG 2006).
There is also the question of statistical power. Are these 14 trials of over 5000 women sufficient to answer the question of effectiveness? In order to detect a 10% improvement in event‐free survival (estimating that 50% of women with early poor prognosis breast cancer will progress without additional treatment by five years, which would decrease to 40% with treatment), it would be necessary to recruit 407 women to each arm of the study with 95% confidence of detecting a difference with 80% power. Therefore, the total number of women in this systematic review is sufficient to detect a difference by five years, and at least one of the individual studies (Dutch Intergp 2003) has the statistical power to detect a 10% difference However, as noted below, five years of follow‐up may not be long enough to reach a conclusion about the relative efficacy of treatments.
Is it appropriate to pool the results of these 14 studies? They randomised women with differing prognoses, in particular with respect to their number of positive lymph nodes. There was also variability in the chemotherapy regimens used. However, inspection of the forest plots shows very little heterogeneity except for three‐year survival outcomes, which were influenced by the differing findings of the smallest trial in the review (MDACC 2000). Sensitivity analysis excluding studies with lower‐risk participants(fewer than 10 nodes) negated the statistically significant benefit shown for high‐dose therapy in event‐free survival rates at three years. This was probably due to reduced power in the meta‐analysis rather than clinical differences in the participants, since there was no statistical heterogeneity and sensitivity analyses allowing for these differences did not change the statistical significance of any other results. Thus it would appear that pooling the results of the studies was appropriate.
Although high‐dose chemotherapy with autograft is associated with considerable morbidity and its role in the treatment of breast cancer has not yet been fully defined, it has often been viewed as a worthwhile treatment for women with poor prognosis or advanced disease (Nieto 2000). As a result, many women in the USA were treated outside of a clinical trial: data from the Autologous Blood and Marrow Transplant Registry suggest that in the USA during the 1990s over 40,000 women with breast cancer received this treatment, yet fewer than 1000 were recruited to clinical trials (ABMTR 2002). This review underscores the need for randomised controlled trials as the only reliable method of establishing effective treatments for women with breast cancer.
Quality of the evidence
Most of the primary studies in this review were at low risk of bias in all but one of the domains assessed. The exception was blinding, which appears unlikely to influence our outcomes of interest. Many of the findings were based on estimated data as study follow‐up was not complete in all women, but findings were largely consistent across trials and it appears unlikely that further data will change the overall finding.
The overall quality of the evidence was high for all comparisons. There was no serious indirectness, inconsistency, imprecision or evidence of publication bias.
However, the ECOG 2003 trialists noted that subgroup analysis excluding women with minor protocol violations showed a longer time to recurrence in the high‐dose group, and they also commented, as did the WSG 2005 trialists, on an apparent late divergence in survival rates. Extended follow‐up will be important to determine whether such differences persist or increase. The Dutch Intergp 2003 trialists suggested that additional follow‐up of five to 10 years might be required before a definitive conclusion about overall survival could be made. We do not plan to update this review unless further compelling evidence emerges.
Potential biases in the review process
The statistical methods used in our review are not ideal, as we have pooled data at specific time points rather than pooling all data to calculate hazard ratios. However as the data mature our findings are consistent with those of the individual patient data meta‐analysis mentioned below (Berry 2011).
Agreements and disagreements with other studies or reviews
The data from 15 RCTs of high‐dose chemotherapy have been combined in an overview and meta‐analysis of individual patient data (Berry 2011). This review included all of the RCTs in our updated review and reached very similar conclusions: "Adjuvant high dose chemotherapy with autologous hematopoietic stem‐cell transplantation prolongs relapse‐free survival in high‐risk primary breast cancer compared with control, but this does not translate into a significant overall survival benefit. Whether high dose chemotherapy benefits patients in the context of targeted therapies is unknown".
Two subsequent systematic reviews (Wang 2012;Pedrazzoli 2015) also reached similar conclusions.
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Forest plot of comparison: 1 High‐dose chemotherapy versus standard chemotherapy, outcome: 1.1 Overall survival.
Forest plot of comparison: 1 High‐dose chemotherapy versus standard chemotherapy, outcome: 1.2 Event‐free survival.
Funnel plot of comparison: 1 High‐dose chemotherapy versus standard chemotherapy, outcome: 1.3 Treatment‐related mortality.
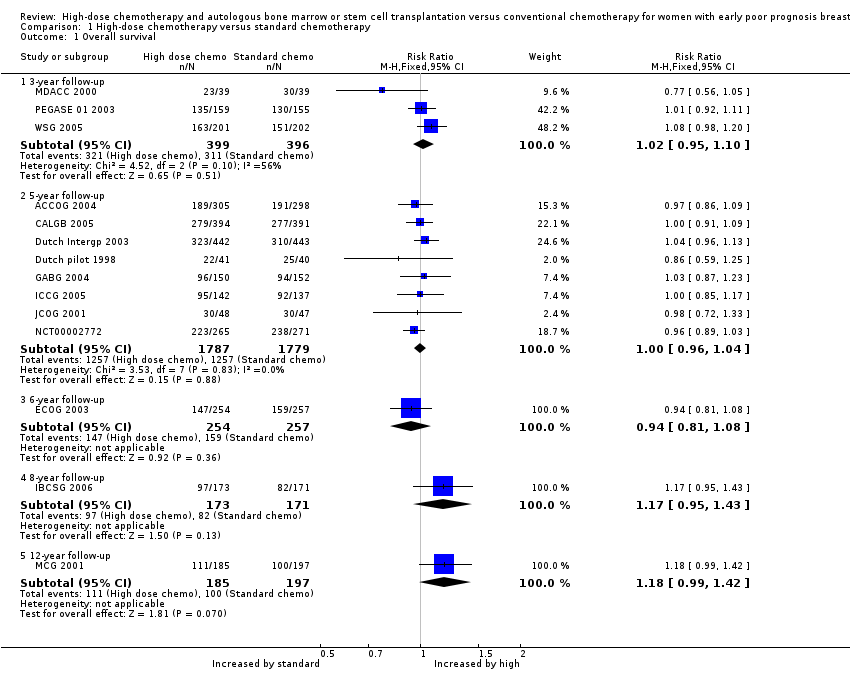
Comparison 1 High‐dose chemotherapy versus standard chemotherapy, Outcome 1 Overall survival.
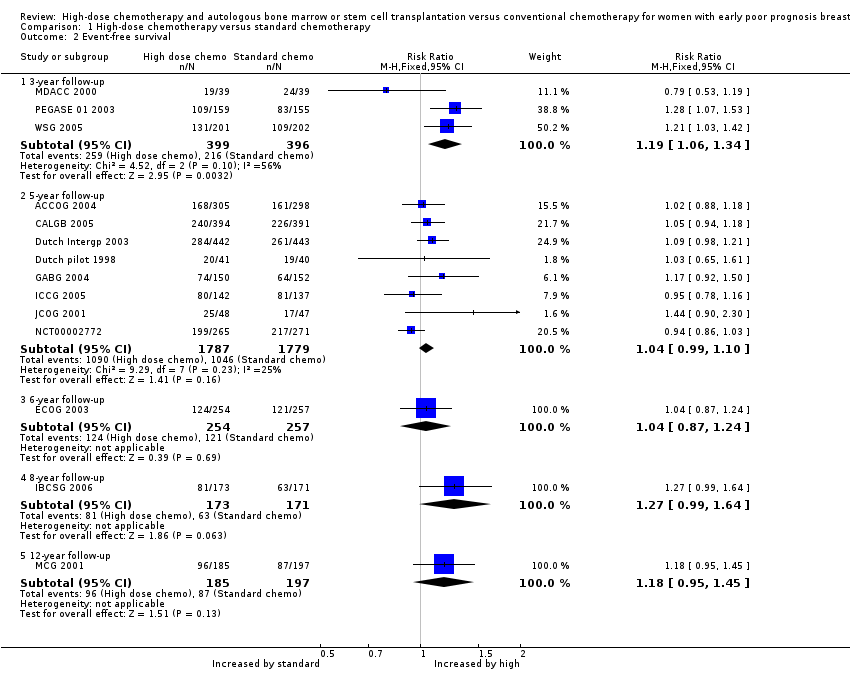
Comparison 1 High‐dose chemotherapy versus standard chemotherapy, Outcome 2 Event‐free survival.

Comparison 1 High‐dose chemotherapy versus standard chemotherapy, Outcome 3 Treatment‐related mortality.

Comparison 1 High‐dose chemotherapy versus standard chemotherapy, Outcome 4 Second cancers.
| High‐dose chemotherapy versus chemotherapy without bone marrow transplant or stem cell rescue | ||||||
| Population: women with early poor prognosis breast cancer | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with standard chemotherapy | Risk with high dose chemotherapy | |||||
| Overall survival at 5‐year follow‐up | 672 per 1000 | 672 per 1000 | RR 1.00 | 3566 (8 RCTs) | ⨁⨁⨁⨁ | |
| Event‐free survival at 5‐year follow‐up | 578 per 1000 | 601 per 1000 | RR 1.04 | 3566 | ⨁⨁⨁⨁ | |
| Treatment‐related mortality | 2 per 1000 | 14 per 1000 | RR 7.97 | 5600 | ⨁⨁⨁⨁ | Most deaths occurred within the first year of treatment |
| Second cancers at 4 ‐ 9‐year median follow‐up | 25 per 1000 | 31 per 1000 | RR 1.25 | 3423 | ⨁⨁⨁⨁ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the median risk in the comparison group and the relative effect of the intervention (and its 95% CI) | ||||||
| GRADE Working Group grades of evidence | ||||||
| Study ID | Median Age | Tumour | Median nodes positive | Minimum nodes positive | > 9 nodes | Oestro positive | Progest. positive | Other | Premenop'sal |
| 45 | 3 cm max. | 9 | 4 | 45% | 31% (ER or PR +ve) | 31% (ER or PR +ve) | 43% receptor unknown | ‐ | |
| 45 | 3 cm median | 14 (range 10 ‐ 52) | 10 | 100% | 69% | ‐ | ‐ | ‐ | |
| 45 | T1 5%; T2 30%; T3 45%; T4 10%; Tx 10% | ‐ | N/A: Had pre‐op chemo | N/A | 20% | 25% | 54% receptor unknown | 83% | |
| 45.7 | T1 22%; T2 60%; T3 16% | ‐ | 4 | 35.8% | 65% | 53% | 28% oestrogen receptor negative | ‐ | |
| 44 | ‐ | ‐ | 10 | ‐ | 60% | 59% | 46% > 14 +ve nodes | 72% | |
| ‐ | ‐ | ‐ | 10 | 100% | 60% | 40% | ‐ | 58% | |
| 46 | T1 26%; T2 51%; T3 20% | 13 | 5 ‐ 10 depending on other prognostic factors | 73% | ‐ | ‐ | 40% oestrogen & progesterone receptor ‐ve | 67% | |
| 47 (range 24 ‐ 60) | T1 28%; T2 54%; T3 14%; unknown 4% | 9 (range 4 ‐ 36) | 4 | 45% | 43% | 25% | 38% receptor status not known | 70% | |
| 46 | ‐ | 16 (range 10 ‐ 49) | 10 | 100% | ‐ | ‐ | ‐ | 74% | |
| 45 | ‐ | ‐ | 10 at diagnosis or 4 after initial chemo | > 60% | 50% | 45% | 5% receptor unknown | 68% | |
| ‐ | ‐ | ‐ | 4 | 62% | ‐ | ‐ | ‐ | ‐ | |
| 46 (mean) | ‐ | 13 | 8 | ? | 31% | ‐ | ‐ | 68% | |
| Not stated. 45% were aged 40 ‐ 49 yrs | 20% had T3 tumour | 8% were N2 | ‐ | ‐ | ‐ | ‐ | 66% ER/PgR +ve; 8% receptor unknown | ‐ | |
| 47 | Mean size 3.3 ‐ 3.5 cm | 17 ‐ 18 | 10 | 100% | 63% | ‐ | ‐ | 53% | |
| +ve = positive | |||||||||
| Stage | What stage means |
| I | Breast tumour 2 cm or less in diameter and does not appear to have spread beyond the breast |
| IIA | Breast tumour over 2 cm in diameter OR has spread to the axillary (underarm) lymph nodes on the same side as the breast cancer. The nodes are not stuck to one another or to the surrounding tissues |
| IIB | Breast tumour over 2 cm in diameter AND has spread to the axillary nodes on the same side as the breast cancer. The nodes are not stuck together or to the surrounding tissues. OR the tumour is larger than 5 cm in diameter (and nodes are clear) |
| IIIA | Breast tumour over 5 cms in diameter AND has spread to the axillary lymph nodes on the same side OR tumour has spread to the lymph nodes on the same side as the breast cancer and the nodes are stuck to each other or to the surrounding tissues |
| IIIB | Breast tumour has spread to chest wall or skin OR tumour has spread to internal mammary lymph nodes on the same side as breast tumour |
| IV | Tumour has spread from breast to distant sites or to supraclavicular (above collarbone) lymph nodes |
| Study | Phase 1 | Phase 2 |
| doxorubicin 75 mg | cyclophosphamide | |
| cyclophosphamide 600 mg | cyclophosphamide 900 mg | |
| cyclophosphamide 500 mg | ‐ | |
| cyclophosphamide 500 mg | ‐ | |
| cyclophosphamide 1400 mg (po) | ‐ | |
| cyclophosphamide 600 mg | cyclophosphamide 1 gm | |
| doxorubicin 60mg or epirubicin 90 mg | cyclophosphamide 1400 mg (po) | |
| cyclophosphamide 600 mg | cyclophosphamide 1200 mg | |
| cyclophosphamide 500 mg | ‐ | |
| epirubicin 120 mg 3 cycles | cyclophosphamide 600 mg | |
| cyclophosphamide 500 mg | ‐ | |
| cyclophosphamide 500 mg | ‐ | |
| sequential administration of 3 cycles each of doxorubicin 80 mg/m², paclitaxel 200 mg/m², and cyclophosphamide | ‐ | |
| cyclophosphamide 600 mg | cyclophosphamide 600 mg |
| Study | Initial phase | High‐dose cycle 1 | High‐dose cycle 2 | High‐dose cycle 3 | High‐dose cycle 4 | Regimen |
| 4 cycles of doxorubicin (as control arm) followed by: | cyclophosphamide 4 gm | cyclophosphamide 6 gm | ‐ | ‐ | Divided doses over 4 days | |
| 4 cycles of cyclophosphamide, doxorubicin and fluorouracil (as control arm) followed by: | cyclophosphamide 5.625 gm | ‐ | ‐ | ‐ | Divided doses over 3 days | |
| 4 cycles of cyclophosphamide, epirubicin and fluorouracil (doses as control arm) followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Divided doses over 4 days | |
| 4 cycles of cyclophosphamide, epirubicin and fluorouracil (as control arm) followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Divided doses over 4 days | |
| 6 cycles of cyclophosphamide, doxorubicin and 5FU (as control arm) followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Continuous infusion over 4 days | |
| 4 cycles of cyclophosphamide and epirubicin (as control arm) followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Divided doses over 4 days | |
| No common path with control group protocol | epirubicin 200 mg | As cycle 1 | As cycle 1 | 3 X 21‐day cycles | ||
| 2 cycles of cyclophosphamide, epirubicin and fluorouracil (as control arm cycles 1 and 2) | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | Continuous infusion over 4 days | |
| 6 cycles of cyclophosphamide, doxorubicin and fluorouracil (as control arm), followed by: | cyclophosphamide 6 gm | ‐ | ‐ | ‐ | ‐ | |
| No common path with control group protocol | cyclophosphamide 7 gm | methotrexate 8gm | epirubicin 120 mg X 2 | thiotepa 600 mg melphalan 160 ‐ 180 mg | 4 high‐dose treatments in sequence | |
| 8 cycles of cyclophosphamide, doxorubicin and fluorouracil (as control arm), followed by: | cyclophosphamide 5.25 gm | As cycle 1 | ‐ | ‐ | Divided doses over 3 days. 2nd cycle given when haematologically safe | |
| 4 cycles of cyclophosphamide, epirubicin and fluorouracil (as control arm), followed by: | cyclophosphamide 120 mg | ‐ | ‐ | ‐ | ‐ | |
| 4 cycles of doxorubicin 80 mg/m² and cyclophosphamide 600 mg/m² (AC) every 3 weeks | STAMP I or STAMP V HDC regimen. STAMP I consisted of cyclophosphamide 1.85 g/m²/d and cisplatin 55 mg/m²/d, followed by carmustine 600 mg/m²; STAMP V consisted of cyclophosphamide 1.5 g/m²/d, carboplatin 200 mg/m²/d, and thiotepa 125 mg/m²/d | ‐ | ‐ | ‐ | ‐ | |
| 2 cycles of cyclophosphamide and epirubicin (as control arm) | cyclophosphamide 3 gm | As cycle 1 | ‐ | ‐ | High‐dose cycles over 28 days |
| Study ID | Data maturity | Median follow‐up |
| No | 4 years | |
| No | 7.3 years | |
| 5 years | 6.9 years | |
| 3 years | 7 years | |
| No | 6.1 years | |
| No | 6.1 years | |
| No | 8.3 years | |
| No | 4.2 years | |
| No | 63 months | |
| 3 years | 11.9 years | |
| No | 11.33 years | |
| 3 years | 3.25 years | |
| No | 5.8 years | |
| 3 years | 4 years |
| Study ID | Haemopoietic | Gastrointestinal | Pulmonary | Cardiac events | Neurological | Other toxicity | Late/ long term | Second cancers | Trialist's summary |
| Standard chemo: Grade 4 neutropenia 15% | Haemorrhage ≥ grade 2: | Nausea ≥ grade 3: | Rhythm toxicity ≥ grade 2: | Cortical neurotoxicity ≥ 1 | Both trial arms: Menopausal symptoms common. | ‐ | ‐ | ‐ | |
| Leukopenia and thrombocytopenia common in both groups but more severe and persistent in HDC arm | ‐ | Toxicity ≥ grade 3: | ‐ | Toxicity ≥ grade 3: | Hepatic toxicity ≥ grade 3: | ‐ | By median 7.5 yrs High‐dose arm: 16 second cancers (4%) (including acute myeloid leukaemia or myelodysplatic syndrome 7; breast cancer 5) | ‐ | |
| High‐dose chemo: all hospitalised for 13 ‐ 30 days for haemopoietic recovery. Median neutropenic fever 5 days Standard chemo: neutropenic fever after 4% of cycles | High‐dose: mucositis 85% (severe in 22%), diarrhoea common. Standard chemo: Mild nausea and vomiting, mucositis (28% of cycles), diarrhoea (4% of cycles) | ‐ | See long‐term events | ‐ | Both arms: alopecia 100%, fatigue common, lymphoedema of arm in 20% High‐dose: ovarian failure 100%, radiation pneumonitis 10%, Standard dose: radiation pneumonitis 2% | High‐dose arm: 1 case hypothyroidism, 1 case auto‐antibody production | At median follow‐up of 7 years: | High‐dose: "Moderately well tolerated but substantial though reversible toxic effects". Standard dose: "Mild toxicity" | |
| High‐dose: transfusion‐dependent 100% | High‐dose: nausea and vomiting 100% | ‐ | High‐dose: cardiac arrhythmia 1/442, possible heart failure 1/442 | ‐ | High‐dose: high fever (necessitating early termination of treatment): 4 women (1%) | ‐ | By median follow‐up 7 years: | High‐dose: "Well tolerated" | |
| High‐dose: leukopenia 98%, granulocytopenia 94%, thrombocytopenia 97%, anaemia 62%, | High‐dose: nausea 32%, vomiting 16%, diarrhoea 22%, stomatitis 37% Standard chemo: nausea 11%, vomiting 8%, stomatitis 4% (all grade 3 or 4) | Standard chemo: 1% (grade 3 or 4) | ‐ | Standard chemo: 6% (grade 3 or 4) | High‐dose: infection 21%, liver effects 13%, skin effects 11%, diabetes 14% Standard dose: hyperglycaemia 2%, phlebitis 1%, hepatotoxicity 1% (all grade 3 or 4) | ‐ | By median 6.1 years: | ‐ | |
| ‐ | High‐dose: Grade 3 or 4 gastrointestinal toxicity < 1%; | Grade 3 or 4 toxicity < 1% | ‐ | High‐dose: Grade 3 or 4 toxicity nil | High‐dose: Grade 3 or 4 toxicity: Bladder < 1%; kidney nil; liver nil | ‐ | ‐ | ‐ | |
| High‐dose: myelosuppression | High‐dose: nausea and vomiting; mucositis | ‐ | ‐ | ‐ | ‐ | Permanent amenorrhoea: High‐dose arm 77/95 (81% overall, age < 40 years 61%; age > 40 years 96%); Standard‐dose arm 61/98 (63% overall age < 40 years 24%; age > 40 years 84%) | By median 8.3 years: | High‐dose: Overall toxicities Grade 3 1%; Grade 4 98%; | |
| High‐dose: leucopenia and thrombocytopenia presumed 100% but nadir count not always available (grade 3 or 4) | High‐dose: nausea and vomiting 46%, mucositis 22% (grade 3 or 4) | High‐dose: Pulmonary embolus 1/143; respiratory failure requiring ventilator 1/143 | High‐dose: severe cardiac arrhythmia 2% (3/143) | ‐ | High‐dose: hair loss 100%, fever (no infection) 17%, infection 24%, "other" 28% (grade 3 or 4), deep vein thrombosis 1/143 | After chemotherapy: 227 toxic events occurred (127 in high‐dose arm, 110 in control arm), of which 30% related to tamoxifen. Of the others, 7 events deemed life‐threatening (5 in high‐dose group, 2 in control arm) | High‐dose: 2/143 (breast 1, ovarian 1) | ||
| High‐dose: All 34 women receiving HDC actually developed grade 4 leukopenia and grade 4 neutropenia; 27 (79%) developed grade 4 and the other 7 grade 3 thrombocytopenia. Standard dose: 7 women (8%) developed grade 4 neutropenia | High‐dose: vomiting 62%, diarrhoea 29%, mucositis 15%, (grade 3 or 4) | ‐ | High‐dose: grade 3 arrhythmia 3%, | ‐ | High‐dose: Grade 3 or 4 infection: 6% | ‐ | ‐ | ‐ | |
| High‐dose: Length of hospital stay not stated. Standard dose: 22% admitted with infection or fever | High‐dose: mild/moderate vomiting 80%, mild/moderate diarrhoea 58%, mild/moderate mucositis 83%. Standard dose: Nausea and vomiting moderate 75%, severe 16%. Diarrhoea moderate 19%, severe 8%. Mucousitis moderate 36%, severe 10% | High‐dose: 1 case (severe) | High‐dose: moderate/severe 8%. Standard dose: 1 woman (1%) had myocardial infarction | High‐dose: hearing loss 2 cases (6%) ‐ 1 permanent, mild/moderate peripheral neuropathy 11% | High‐dose: Renal: 25% (22% mild, < 3% severe), hepatic (mild/moderate) 31%, bladder (moderate) 25%, skin (mild) 8% | High‐dose: 1 case of avascular necrosis | High‐dose: 1 case of acute myeloid leukaemia | "Overall there was greater and more frequent morbidity associated with high dose chemotherapy" | |
| ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| High‐dose: 62% had haematologic toxicity during induction and 92% had it during transplantation. 3 women had myelodysplastic syndrome Controls: 59% had haematologic toxicity 2 women had myelodysplastic syndrome | ‐ | ‐ | ‐ | ‐ | ‐ | High‐dose: 44% of women experienced grade 3 or 4 nonhaematologic toxicity during induction while 80% experienced grade 3 or 4 nonhematologic toxicity during transplantation. Control arm: Approximately 63% experienced grade 3 or 4 nonhaematologic toxicity, most commonly fatigue, nausea and vomiting, infection, febrile neutropenia, mucositis, and sensory neuropathy | ‐ | High‐dose: 44% had | |
| ‐ | High‐dose arm: nausea 25%, mucositis 18%, diarrhoea 5% | High‐dose arm: 1% | High‐dose arm: 3% | ‐ | High‐dose arm: grade 3 or 4 skin toxicity 3%, amenorrhoea 100% | ‐ | ‐ | Both high‐dose chemotherapy and dose‐dense conventional chemotherapy are feasible with tolerable toxicity in a multicentre setting |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Overall survival Show forest plot | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 3‐year follow‐up | 3 | 795 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.95, 1.10] |
| 1.2 5‐year follow‐up | 8 | 3566 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.96, 1.04] |
| 1.3 6‐year follow‐up | 1 | 511 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.81, 1.08] |
| 1.4 8‐year follow‐up | 1 | 344 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.95, 1.43] |
| 1.5 12‐year follow‐up | 1 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.99, 1.42] |
| 2 Event‐free survival Show forest plot | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 3‐year follow‐up | 3 | 795 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [1.06, 1.34] |
| 2.2 5‐year follow‐up | 8 | 3566 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.99, 1.10] |
| 2.3 6‐year follow‐up | 1 | 511 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.87, 1.24] |
| 2.4 8‐year follow‐up | 1 | 344 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.99, 1.64] |
| 2.5 12‐year follow‐up | 1 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.95, 1.45] |
| 3 Treatment‐related mortality Show forest plot | 14 | 5600 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.97 [3.99, 15.92] |
| 4 Second cancers Show forest plot | 7 | 3423 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.90, 1.73] |
| 4.1 By median 4‐ to 5‐year follow‐up | 2 | 817 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.34 [0.61, 8.99] |
| 4.2 By median 6‐year follow‐up | 1 | 511 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [0.75, 3.78] |
| 4.3 By median 7‐year follow‐up | 3 | 1751 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.69, 1.51] |
| 4.4 By median 8‐ to 9‐year follow‐up | 1 | 344 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.97 [0.61, 14.49] |