Inhibiteurs des canaux calciques pour le phénomène de Raynaud primitif et secondaire
Résumé scientifique
Contexte
Le phénomène de Raynaud est une maladie vasospastique caractérisée par une pâleur, une cyanose des doigts et une douleur des extrémités. Le phénomène de Raynaud primitif n’est pas associé à une maladie sous‐jacente, tandis que le phénomène de Raynaud secondaire est associé à des maladies du tissu conjonctif telles que la sclérodermie systémique, le lupus érythémateux disséminé, et les connectivites mixtes. Les inhibiteurs des canaux calciques favorisent la vasodilatation et sont couramment utilisés lorsqu’un traitement médicamenteux est nécessaire dans le phénomène de Raynaud.
Objectifs
Évaluer les bénéfices et les risques des inhibiteurs des canaux calciques (ICC) par rapport à un placebo pour le traitement des personnes atteintes de phénomène de Raynaud, en fonction du type de celui‐ci (primitif ou secondaire) ainsi que du type et de la dose d’ICC.
Stratégie de recherche documentaire
Nous avons effectué des recherches dans le registre Cochrane des essais contrôlés (19 mai 2017), MEDLINE (de 1946 au 19 mai 2017), Embase (de 1947 au 19 mai 2017), clinicaltrials.gov et le Système d'enregistrement international des essais cliniques (ICTRP) de l’Organisation mondiale de la Santé (OMS). Nous n'avons appliqué aucune restriction concernant la langue. Nous avons également consulté les références bibliographiques des articles identifiés et contacté des experts pour obtenir des données supplémentaires non publiées.
Critères de sélection
Tous les essais contrôlés randomisés (ECR) comparant des inhibiteurs calciques à un placebo.
Recueil et analyse des données
Deux auteurs de la revue ont évalué de façon indépendante les résultats de la recherche et le risque de biais des essais et extrait les données. Nous avons utilisé l’approche GRADE pour évaluer la qualité des preuves.
Résultats principaux
Cette revue contient 38 ECR (33 ECR en cross‐over) d’une durée moyenne de 7,4 semaines et portant sur 982 participants ; cependant, tous les essais n’ont pas rapporté tous les critères de jugement d'intérêt. Neuf des essais identifiés portaient sur des patients présentant un phénomène de Raynaud primitif (N = 365), cinq sur des patients porteurs d’un phénomène de Raynaud secondaire (N = 63), et les essais restants évaluaient un ensemble de patients atteints de phénomène de Raynaud primitif ou secondaire (N = 554). Les types de risque de biais les plus fréquents étaient des données incomplètes concernant les critères de jugement, la faible notification de la randomisation et des méthodes d’assignation.
Lorsque les chercheurs ont évalué à la fois le phénomène de Raynaud primitif et secondaire, des preuves de qualité modérée (abaissée du fait de leur incohérence) issues de 23 essais et portant sur 528 participants ont indiqué que les inhibiteurs des canaux calciques (ICC) étaient plus efficaces que le placebo pour réduire la fréquence des crises. Les ICC permettaient d'obtenir une réduction du nombre moyen de crises, avec six crises de moins par semaine (différence moyenne pondérée (DMP) de ‐6,13, intervalle de confiance (IC) à 95 % de ‐6,60 à 5,67 ; I² = 98 %) par rapport à 13,7 crises par semaine sous placebo. Lorsque les auteurs de la revue ont exclu l’étude Kahan 1985C, un essai montrant une importante diminution de la fréquence des crises, les données ont montré que les ICC permettaient d'obtenir une réduction de la fréquence des crises avec 2,93 crises de moins par semaine (IC à 95 % de ‐3,44 à ‐2,43 ; I² = 77 %).
Des preuves de mauvaise qualité (abaissée en raison de leur imprécision et de leur incohérence) issues de 6 essais et portant sur 69 participants suggèrent que la durée moyenne des crises ne différait pas de façon statistiquement significative ou cliniquement significative entre les ICC et un placebo (DMP de ‐1,67 minutes, IC à 95 % de ‐3,29 à 0) ; ce qui équivaut à une différence de ‐9 % (IC à 95 % de ‐18 % à 0 %).
Des preuves de qualité modérée (abaissée en raison de leur incohérence) issues de 16 essais et portant sur 415 participants ont montré que les ICC réduisaient la gravité des crises de 0,62 cm (IC à 95 % de ‐0,72 à 0,51) sur une échelle visuelle analogique de 10 cm (les scores les plus bas indiquant une sévérité moindre), ce qui correspond à une réduction absolue en pourcentage de 6 % (IC à 95 % de ‐11 % à ‐8 %) et relative de 9 % (IC à 95 % de ‐11 % à ‐8 %), ce qui n’est probablement pas cliniquement significatif.
L'amélioration de la douleur (preuves de mauvaise qualité, abaissée en raison de leur imprécision et de leur incohérence) et de l’incapacité mesurée par une évaluation globale du patient (preuves de qualité modérée, abaissée à cause de leur imprécision) était en faveur des ICC (douleur : DMP de ‐1,47 cm, IC à 95 % de ‐2,21 à ‐0,74 ; évaluation globale du patient : DMP de ‐0,37 cm, IC à 95 % de ‐0,73 à 0, pour l’évaluation sur une échelle visuelle analogique de 0 à 10 cm, les scores les plus bas indiquant une douleur et une incapacité moindres). Cependant, ces estimations n’avaient probablement pas une puissance statistique suffisante car elles étaient basées sur un nombre limité de participants (respectivement 62 et 92). Pour l’évaluation de la douleur, l'amélioration absolue en pourcentage était de 15 % (IC à 95 % de ‐22 % à ‐7 %) et l’amélioration relative de 47 % (IC à 95 % de ‐71 % à ‐24 %). Pour l'évaluation globale du patient, l'amélioration absolue en pourcentage était de 4 % (IC à 95 % de ‐7 % à 0 %) et l’amélioration relative de 9 % (IC à 95 % de ‐19 % à 0 %).
Les analyses en sous‐groupes par type de phénomène de Raynaud, classe d’ICC et dose d’ICC suggèrent que les ICC de la famille des dihydropyridines, à forte dose, pourraient être plus efficaces sur le syndrome de Raynaud primitif que sur la forme secondaire, et que les ICC ont probablement un effet plus important sur la forme primitive que sur la forme secondaire. Toutefois, les différences étaient faibles et n’ont pas été observées pour tous les critères de jugement. Les dihydropyridines ont été étudiées car il s’agit d’un sous‐groupe d’ICC non cardiosélectifs, traditionnellement utilisés dans le traitement du phénomène de Raynaud, tandis que d’autres ICC tels que le vérapamil ne sont pas couramment utilisés et que le diltiazem n'est pas utilisé en première ligne. La plupart des données des essais portaient sur la nifédipine.
Les arrêts prématurés des études en raison d’effets indésirables étaient peu concluants en raison d’un large intervalle de confiance (risque relatif [RR] 1,30, IC à 95 % de 0,51 à 3,33) obtenu à partir de deux études en parallèle portant sur 63 participants (preuves de faible qualité, rabaissée en raison de l’imprécision et d’un taux d’attrition élevé) ; les différences absolues et relatives en pourcentage pour les arrêts prématurés étaient de 6 % (IC à 95 % de ‐14 % à 26 %) et 30 % (IC à 95 % de ‐49 % à 233 %), respectivement. Bien qu’il n'y ait pas eu de méta‐analyse, les arrêts prématurés étaient plus fréquents avec les ICC qu’avec le placebo dans les essais en cross‐over. Les effets secondaires les plus fréquents étaient des céphalées, des étourdissements, des nausées, des palpitations et des œdèmes des chevilles. Cependant, aucune étude n'a rapporté d'événement indésirable grave (décès ou hospitalisation).
Conclusions des auteurs
Des essais contrôlés randomisés, avec des preuves de qualité faible à modérée, ont montré que les ICC (en particulier la classe des dihydropyridines) pourraient être utiles pour réduire la fréquence, la durée, la sévérité des crises, ainsi que la douleur et l’incapacité associées au phénomène de Raynaud. Des doses élevées pourraient être plus efficaces que des doses plus faibles et ces ICC pourrait être plus efficaces sur le phénomène de Raynaud primitif. Bien qu'il y ait eu davantage d’arrêts prématurés en raison d’événements indésirables dans les groupes avec traitement, aucun événement indésirable grave n’a été rapporté.
PICOs
Résumé simplifié
Inhibiteurs des canaux calciques pour le traitement des patients souffrant du phénomène de Raynaud
Le phénomène de Raynaud résulte d'une diminution du flux sanguin vers les doigts et les orteils causée par un vasospasme. Les symptômes incluent une décoloration (extrémité des doigts devenant blanche, puis bleue et/ou rouge), une douleur et, dans certains cas graves, des ulcérations des doigts. Le froid, le stress et l’anxiété sont les facteurs déclenchants les plus fréquents d’une crise du phénomène de Raynaud. Le phénomène de Raynaud primitif n’est associé à aucune maladie sous‐jacente, à la différence de la forme secondaire qui peut être liée, par exemple, à une sclérodermie systémique.
Cette revue a évalué les bénéfices et les risques des inhibiteurs des canaux calciques (ICC, inhibiteurs calciques) comparativement à un placebo (une substance ayant le même aspect que le médicament actif mais ne contenant aucun principe actif) pour le traitement des patients atteints du phénomène de Raynaud, sur la base des études publiées jusqu’au 19 mai 2017. Les inhibiteurs calciques sont des médicaments qui augmentent le débit sanguin vers les doigts et qui sont généralement utilisés comme traitement de première ligne chez les patients souffrant du phénomène de Raynaud. L’objectif de cette revue était d'évaluer les bénéfices et les effets délétères des inhibiteurs calciques de façon globale, ainsi qu’en fonction de la dose, du type de médicament et du type de phénomène de Raynaud (primitif vs secondaire).
Caractéristiques de l’étude
Nous avons identifié et inclus 38 études portant sur 982 personnes âgées de 18 ans et plus, et souffrant de ce trouble depuis plus ou moins longtemps et avec une intensité variable. Neuf études incluaient des patients atteints de phénomène de Raynaud primitif, cinq études incluaient des patients atteints de phénomène de Raynaud secondaire et les études restantes évaluaient des patients présentant les deux types. La durée des essais variait entre 2 et 20 semaines.
Qu’apporte cette revue concernant l’utilisation des ICC par rapport au placebo dans le phénomène de Raynaud ?
Les auteurs de la revue ont trouvé que :
• les ICC réduisent probablement légèrement la fréquence, la gravité et la perception globale des patients des crises du phénomène de Raynaud (preuves de qualité moyenne, abaissée en raison d’imprécisions et d’incohérences) ;
• les ICC pourraient améliorer légèrement la douleur et la durée des crises du phénomène de Raynaud (preuves de faible qualité, abaissée en raison d’imprécisions et d’incohérences) ;
• compte tenu du manque de données et des taux d'abandon élevés, les effets des ICC concernant le risque d’abandon en raison d’effets secondaires du traitement restent incertains ;
• les effets secondaires les plus courants étaient des céphalées, des étourdissements, des nausées, des palpitations, et des œdèmes des chevilles ;
• aucun événement indésirable grave (décès ou hospitalisation) n’a été rapporté.
Meilleure estimation de l'effet des ICC pris pendant 2 à 20 semaines par des personnes atteintes de phénomène de Raynaud
Lorsque les investigateurs ont évalué le phénomène de Raynaud à la fois primitif et secondaire, ils ont rapporté que les 528 personnes ayant pris des ICC avaient présenté 6 crises de moins par semaine que celles ayant pris un placebo. Les personnes ayant pris un ICC avaient en moyenne 8 crises par semaine, contre 14 chez celles prenant un placebo.
La durée des crises (en minutes) était pratiquement la même chez les sujets ayant pris un ICC ou un placebo. Cependant, ce résultat est basé sur un petit nombre de personnes.
La sévérité des crises, mesurée sur une échelle visuelle analogique de 10 cm (faibles scores = faible sévérité des crises) était inférieure de 0,62 cm avec les ICC, ce qui équivaut à une réduction de 6 %. Les personnes ayant pris un ICC évaluaient la gravité d'une crise à 6,1 cm contre 6,7 cm pour les personnes sous placebo.
La douleur a été réduite de 1,5 points sur une échelle de 0 à 10 (réduction absolue de 15 %, un score plus bas équivalant à une douleur moindre) avec les ICC comparés au placebo. Les personnes ayant pris un ICC ont rapporté un score de douleur de 1,6 points contre 3,1 points pour les patients sous placebo.
L’incapacité globale a été réduite de 0,4 points sur une échelle de 0 à 10 (réduction absolue de 4 %, un score plus bas équivalant à une incapacité moindre) chez les personnes ayant pris des ICC par rapport au placebo. Les personnes ayant pris un ICC ont rapporté un score d’incapacité de 3,5 points contre 3,9 points pour les patients sous placebo.
Six personnes de plus parmi 100 personnes abandonnaient l'étude en raison d'effets indésirables (6 % d'arrêts prématurés en plus chez les personnes ayant pris des ICC). Sur 100 personnes prenant un ICC, 25 abandonnaient l’étude, contre 19 sur 100 sous placebo.
Cette revue suggère que les ICC (en particulier les médicaments appartenant à la classe des dihydropyridines comme la nifédipine) à haute dose pourraient être bénéfiques pour le traitement du phénomène de Raynaud, en particulier primitif. Bien qu’un nombre légèrement plus grand de participants sous ICC aient arrêté le traitement en raison d’effets secondaires, aucun effet secondaire grave n’a été rapporté.
Authors' conclusions
Summary of findings
| Calcium channel blockers (CCBs) compared with placebo for treatment of Raynaud's phenomenon | ||||||
| Patient or population: patients with Raynaud's phenomenon | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | CCBs (all) | |||||
| Frequency of attacks | Mean frequency of attacks in control groups: 13.7 attacksa | Mean frequency of attacks in intervention groups: 6.13 lower (6.60 to 5.67 lower)b | 528 | ⊕⊕⊕⊝ moderatec | Note: Excluding a study with a very large reduction in frequency of attacks changed the mean difference to ‐2.93 per week (95% CI ‐3.44 to ‐2.43). Absolute risk difference: N/Ad Relative percent change: ‐44% (95% CI ‐48% to ‐41%) | |
| Duration of attacks Average duration per attack measured in minutes | Mean duration of attacks in control groups: | Mean duration of attacks in intervention groups: (‐3.29 to 0) | 69 | ⊕⊕⊝⊝ lowc,e | NNTB: N/Ad Absolute risk difference: N/Ad Relative percent change: ‐9% (95% CI ‐18% to 0%) | |
| Severity of attacks | Mean severity of attacks in control groups: | Mean severity of attacks in intervention groups: | 415 | ⊕⊕⊕⊝ moderatec | NNTB: N/Ad Absolute risk difference: ‐6% (95% CI ‐7% to ‐5%) Relative percent change: ‐9% (95% CI ‐11% to ‐8%) | |
| Pain Average pain per attack, measured on a 10‐cm visual analogue scale (0 = no pain, 10 = maximal pain) | Mean pain in control groups: | Mean pain in intervention groups: | 62 | ⊕⊕⊝⊝ lowc,e | N/Ad Absolute risk difference: ‐15% (95% CI ‐22% to ‐7%) Relative percent change: ‐47% (95% CI ‐71% to ‐24%) | |
| Patient global Follow‐up: 5 weeks | Mean patient global in control group: 3.9 cma | Mean patient global in intervention groups: | 92 | ⊕⊕⊕⊝ moderatee | NNTB: N/Af Absolute risk difference: ‐4% (95% CI ‐7% to 0%) Relative percent change: ‐9% (95% CI ‐19% to 0%) | |
| Number of withdrawals due to adverse events | 194 per 1000 | 252 per 1000 | RR 1.30 | 63 | ⊕⊕⊝⊝ lowe,g | NNTH: N/Af Absolute risk reduction: 6% (95% CI ‐14% to 26%) Relative percent change: 30% (95% CI ‐49% to 233%) |
| Serious adverse events | See comment. | See comment. | Not estimable | 0 | See comment. | No serious adverse events reported |
| *The basis for the assumed risk (eg, median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aFinal value: weighted mean of scores in placebo group across studies in the meta‐analysis. | ||||||
Background
Description of the condition
Raynaud's phenomenon (RP) is defined as vasospasms or "attacks" of the arteries or arterioles of the extremities (and rarely other areas) causing pallor and at least one other color change upon reperfusion, such as cyanosis or rubor (Herrick 2005; Wigley 2002). During an RP attack, blood flow to the extremities is restricted with subsequent pallor and/or pain. Each attack is characterized by its frequency, duration, and level of pain. Primary RP is idiopathic and occurs in the absence of other underlying causes such as connective tissue disease. Secondary RP occurs in people with underlying diseases affecting the blood vessels, especially systemic sclerosis (also called SSc, scleroderma) and systemic lupus erythematosus (Wigley 2002). RP may also be accompanied by digital ulcers, which may occur secondary to severe ischemia (loss of blood to the digits) (Wigley 2002). In most cases (> 80%), exposure to cold temperatures triggers this physiologic response, but emotional stress has been documented as another trigger (García‐Carrasco 2008; Wigley 2002). Primary RP has an earlier onset (median age at onset is around 14 years) and is characterized by milder symptoms. Secondary RP often has a later onset (usually after age 40) with more severe symptoms and may be associated with complications such as tissue loss, ulcers, and amputation (Wigley 2002). Irreversible digital ischemia may develop in patients whose RP occurs secondary to a systemic sclerosis spectrum disorder (Herrick 2005).
The prevalence of this condition is based on climate conditions so is geographically variable but has been estimated at around 3% to 5% in the general population, with most cases (80% to 90%) diagnosed as primary RP (Maundrell 2015; Silman 1990). Secondary RP accounts for about 10% to 20% of RP prevalence, but this depends on the underlying cause. More than 90% of patients with SSc have RP (Levien 2010; Maundrell 2015). SSc is an autoimmune connective tissue disease characterized by fibrosis of the skin and internal organs, including the gastrointestinal tract, lungs, kidney, and heart, along with significant vasculopathy and pulmonary arterial hypertension (Ortonne 1989). In general, RP is much more common among women (prevalence of primary RP has been estimated at between 2% and 20% in women compared with 1% to 12% among men), and primary RP has a genetic component (about 50% of patients with primary RP have a first‐degree relative with the disease) (Maundrell 2015). Diagnosis of primary RP is based on patient history (i.e., sensitivity to cold exposure with pallor, then rubor or cyanosis of the fingers and toes after cold exposure) and a thorough evaluation to rule out the presence of underlying causes. Diagnosis of secondary Raynaud's phenomenon may be associated with older age at onset (i.e., after age 40), often with more severe symptoms, as well as positive laboratory tests, suggesting an underlying connective tissue disease (i.e., positive antinuclear antibodies, positive rheumatoid factor, and the presence of specific autoantibodies), and magnification of the nail folds, indicating the presence of a microvascular disease (Wigley 2002).
The pathogenesis of Raynaud's phenomenon is not clearly understood, but a general hypothesis is that the major underlying cause is an imbalance of vasoconstrictors and vasodilators (with imbalance more toward the prevalence of vasoconstrictors) (Herrick 2005). Existing evidence suggests that causes of the underlying pathogenesis of Raynaud's phenomenon likely include abnormalities in the blood vessels (i.e., smooth muscle and endothelium), in neural control of vascular tone, and in intravascular mediators, including those produced by platelet activation and oxidative stress (Herrick 2005). However, vascular abnormalities may be minimal in primary Raynaud's phenomenon and more severe in secondary Raynaud's phenomenon (Herrick 2005; Wigley 2002). This might explain why Raynaud's phenomenon secondary to systemic sclerosis spectrum disorders but not primary Raynaud's phenomenon often leads to irreversible digital ischemia and is much more severe (Herrick 2005). In addition, secondary Raynaud's phenomenon is often associated with structural abnormalities in the microvascular system and arteries. Given that Raynaud's phenomenon is more common among women, some have hypothesized that hormonal factors may be involved (Herrick 2005). It has been estimated that 14% to 37% of cases of primary Raynaud's phenomenon eventually progress to secondary Raynaud's phenomenon (Maundrell 2015).
For most people with Raynaud's phenomenon (the vast majority who have primary Raynaud's phenomenon), treatment is conservative (i.e., avoiding cold temperatures and emotional stress, keeping warm, stopping smoking); however, for RP requiring a pharmacological intervention (usually secondary RP), many different drugs may be beneficial (García‐Carrasco 2008; Goundry 2012; Herrick 2005; Wigley 1987).
Description of the intervention
Many randomized controlled trials have examined treatments for both primary and secondary RP. Secondary RP has been studied mostly in SSc and other connective tissue diseases. Conservative treatments and older treatments (i.e. ganglion blockers, alpha blockers) have been superseded by a variety of drugs considered to be more efficacious with lower side effect profiles (Hansteen 1976). These include calcium channel blockers (CCBs), prostacyclin analogues, angiotensin‐converting enzyme inhibitors, and phosphodiesterase inhibitors, among others (García‐Carrasco 2008). CCBs have become the first line of pharmacological treatment for RP owing to their effectiveness and tolerability (García‐Carrasco 2008).
How the intervention might work
Calcium channel blockers (CCBs) are calcium channel antagonists that bind to voltage‐gated calcium channels to prevent influx of calcium ions into smooth and cardiac muscle cells, thereby promoting vasodilation (Sturgill 1998). The most common class of CCBs is the dihydropyridines, which include nifedipine, nicardipine, amlodipine, and felodipine. Non‐dihydropyridine classes of CCBs include benzothiazepine (i.e., diltiazem), phenylalkylamine (i.e., verapamil), and others. Dihydropyridines are more effective for RP, as they are highly selective for vascular smooth muscle in the walls of arteries. They are fast acting and thus are used more often (Sturgill 1998).
Why it is important to do this review
Multiple studies have shown that CCBs have some efficacy in treating individuals with RP. Previous meta‐analyses showed efficacy in the treatment of patients with primary RP and RP secondary to SSc (Ennis 2016; Thompson 2001; Thompson 2005). However, the vascular dysfunction that underlies RP is not clearly understood, and variable patient responses to treatment are based on RP type and severity. As such, no specific guidelines outlining the most efficacious drug interventions have been developed (Dziadzio 1999; Goundry 2012). Moreover, the nuances of treatment have not been well studied. No previous meta‐analyses have examined effects of dose or CCB type, or differences in response dependent on the subtype of RP. This review is different from previous reviews in that review authors analyzed effect of CCBs in both primary and secondary RP and by CCB type and dose.
This review is based on the generic protocol for drug interventions for Raynaud's phenomenon (Pope 2015).
Objectives
To assess the benefits and harms of calcium channel blockers (CCBs) versus placebo for treatment of individuals with Raynaud’s phenomenon (RP) with respect to Raynaud's type (primary vs secondary) and type and dose of CCBs.
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs) and cross‐over RCTs that lasted one week or longer. We included studies reported as full text, those published as abstract only, and unpublished data. We applied no language restrictions.
Types of participants
We used no standardized definition of RP. Studies included primary RP and/or RP secondary to systemic sclerosis or other connective tissue diseases (LeRoy 1992; Masi 1980) and enrolled participants with RP at any stage.
In the absence of an accepted definition for RP, we included all participants reported to have RP and noted the criteria that authors used to define RP.
Types of interventions
Interventions of interest were CCBs and placebo. We applied no restrictions on interventions and comparators, such as delivery, dose, duration, and intensity. We allowed all co‐interventions.
Types of outcome measures
We considered outcomes for trials one week or longer in duration. Outcome measurements included the following.
Major outcomes
-
Frequency of attacks (average attacks/week)
-
Duration of attacks (average duration per attack in minutes)
-
Severity of attacks
-
Pain (i.e. visual analogue scale [VAS], numerical rating scale [NRS])
-
Patient global assessment (measured on various scales, i.e. 0 to 10 VAS)
-
Withdrawals (due to treatment adverse effects)
-
Serious adverse events (treatment adverse effects leading to death or withdrawal from study and hospitalization)
Minor outcomes
-
Function
-
Raynaud's condition score (RCS; Merkel 2002)
-
Physician's global assessment
-
Change in digital ulceration
-
Treatment preference
-
General improvement
-
Side effects
Search methods for identification of studies
Electronic searches
We designed a sensitive search strategy to retrieve RCTs from electronic bibliographic databases. We identified items from the following databases on May 19, 2017.
-
Cochrane Library via Wiley (May 19, 2017) including the Cochrane Central Register of Controlled Trials (CENTRAL), the Database of Reviews of Effects (DARE), Health Technology Assessment (HTA), and the Economic Evaluations Database (EED).
-
MEDLINE via OVID (1946 to May 19, 2017).
-
Embase via OVID (1947 to May 19, 2017).
-
Clinicaltrials.gov (all years).
-
World Health Organization (WHO) International Clinical Trials Portal (all years).
We applied no language restrictions. We used the randomized controlled trials filter from Chapter 6 of the Cochrane Handbook for Systematic Reviews of Interventions. We devised the search strategy for the Cochrane Library Web interface, then adapted it for use with other databases. We have presented the search strategy in Appendix 1.
Searching other resources
We searched all bibliographies of retrieved articles and contacted key experts for additional and unpublished data.
For safety assessments, we searched the websites of regulatory agencies including US Food and Drug Administration‐MedWatch (http://www.fda.gov/Safety/MedWatch/default.htm), European Medicines Evaluation Agency (http://www.ema.europa.eu), Australian Adverse Drug Reactions Bulletin (http://www.tga.gov.au/safety/ews‐monitoring.htm), and UK Medicines and Healthcare products Regulatory Agency (MHRA) pharmacovigilance and drug safety updates (http://www.mhra.gov.uk/Safetyinformation/index.htm), using the keywords "nifedipine," "nicardipine," "nisoldipine," "diltiazem," "verapamil," "amlodipine," "isradipine," and "BAY K 9320," on July 23, 2017.
On July 19, 2017, we searched PubMed for errata or retractions from included studies published in full text (www.ncbi.nlm.nih.gov/pubmed) and found none.
Data collection and analysis
Selection of studies
We included only randomized trials that compared CCBs versus placebo. We included trials comparing CCBs versus other active treatments if they also included a placebo group. We excluded trials comparing CCBs versus other forms of treatment (i.e., natural herbal methods and surgical methods). Two review authors (PTi, SH, or FR) independently reviewed references retrieved through the search and identified studies that met the inclusion criteria. JP resolved differences regarding selection.
Data extraction and management
We recorded study characteristics and outcome data on a data collection form that had been piloted on at least one study in the review. One review author (FR) extracted study characteristics from included studies, and a second review author (LJM or ETG) spot‐checked study characteristics for accuracy against the trial report. We extracted the following study characteristics.
-
Methods: study design, total duration of study, details of any "run‐in" period, washout period, number of study centers and locations, study setting, withdrawals, and date of study.
-
Participants: N, mean age, age range, sex, disease duration, severity of condition, diagnostic criteria, inclusion criteria, and exclusion criteria.
-
Interventions: intervention, comparison, concomitant medications, and excluded medications.
-
Outcomes: primary and secondary outcomes specified and collected and time points reported.
-
Characteristics of trial design: as outlined below in the Assessment of risk of bias in included studies section.
-
Notes: funding for trial and notable declarations of interest of trial authors.
We extracted the number of events and the number of participants per treatment group for dichotomous outcomes, and means and standard deviations and number of participants per treatment group for continuous outcomes. We converted the standard deviation (SD) to a standard error (SE) for entry into the generic inverse variance method in RevMan version 5 (2014), using the formula SE = SD/(square root) (N).
We noted in the Characteristics of included studies table if outcome data were not reported in a usable way, and when data were transformed or estimated from a graph. We resolved disagreements by consensus or by consultation with a third person (JP or GAW). One review author (FR) transferred data into the Review Manager RevMan version 5 (2014). We double‐checked that data were entered correctly by comparing data presented in the systematic review against data in the study reports.
For all outcomes, we extracted data from studies and reported as follows:
-
If both final values and change from baseline values were reported for the same outcome, we preferentially extracted changes from baseline.
-
If both unadjusted and adjusted values were reported for the same outcome, we extracted the adjusted values.
-
If data are analyzed based on intention‐to‐treat (ITT) and another sample (i.e. per‐protocol, as‐treated), we extracted both but noted these differences.
-
If data were available for multiple time points, we used the data that corresponded most closely with those of other RCTs.
For cross‐over studies, we extracted changes in each arm (placebo and treatment) by comparing them with baseline values for each arm when available, or by noting differences between final treatment and placebo.
Assessment of risk of bias in included studies
Two review authors (SH, PTi, or FR) independently assessed the risk of bias of each included study according to the domain‐based evaluation outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We compared assessments, identified inconsistencies, and reached consensus.
For each included study, we rated the following domains as "low risk," "high risk," or "unclear risk" for "risk of bias" assessments.
-
Random sequence generation.
-
Allocation concealment.
-
Blinding of participants and personnel.
-
Blinding of outcome assessment.
-
Incomplete outcome data.
-
Selective outcome reporting.
-
Other bias: We considered cross‐over effects and baseline characteristics.
We reported an assessment of bias for each included study in a "Risk of bias" table within the Characteristics of included studies section.
Measures of treatment effect
We estimated treatment effects for continuous outcomes using weighted mean difference (WMD) or standardized mean difference (SMD). When investigators used different scales to measure the same conceptual outcome (i.e. pain), we calculated SMDs instead, along with corresponding 95% confidence intervals (CIs). We back‐translated the SMD to a typical scale (i.e. 0 to 10 for pain) by multiplying the SMD by a typical among‐person standard deviation (i.e. standard deviation at baseline of the control group from the most representative trial) (as per Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions) (Schünemann 2011).
We estimated dichotomous outcomes using risk ratios (RRs) by dividing the proportion of events in the treatment group by the proportion of events in the control group. We reported 95% CIs with each outcome estimate.
In the Effects of interventions section under Results and in the "Comments" column of summary of findings Table for the main comparison, we provided absolute percent difference, relative percent change from baseline, and number needed to treat for an additional beneficial outcome (NNTB) (we provided NNTB only when the outcome showed a statistically significant difference).
For dichotomous outcomes, we calculated the NNTB from the control group event rate and the risk ratio using the Visual Rx NNT calculator (Cates 2008). We calculated the NNTB using the Wells calculator (available at the Cochrane Musculoskeletal Group [CMSG] Editorial Office).
For dichotomous outcomes, we calculated absolute risk difference using the risk difference statistic in RevMan version 5 (2014) and expressed the result as a percentage. For continuous outcomes, we calculated absolute benefit as improvement in the intervention group minus improvement in the control group, in original units, expressed as a percentage.
We calculated relative percent change for dichotomous data as "Risk ratio ‐ 1" and expressed this as a percentage. For continuous outcomes, we calculated relative difference in changes from baseline as absolute benefit divided by the baseline mean of the control group, expressed as a percentage.
Unit of analysis issues
The participant was the unit of analysis for each outcome. For cross‐over trials, calculation of standard errors and use of the generic inverse variance method accounted for the fact that observations were paired.
When a single trial reported multiple trial arms, we included only the relevant arms. If two comparisons were combined in the same meta‐analysis (i.e. different doses, different drugs), we halved the control group to avoid double‐counting.
Dealing with missing data
We made all possible efforts to obtain any missing data. We had foreign language studies translated when possible, and we used the Cochrane network to try to obtain articles that were not available at the national libraries in Ottawa and Washington, if they also were not available at local universities. When feasible, we estimated missing standard deviations (using standard errors, P values, confidence intervals, error bars in graphs, range and sample size, etc., if available).
Assessment of heterogeneity
We assessed heterogeneity by using Chi² and I² tests and by visually inspecting forest plots for outliers.
As recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011), an I² value of 0% to 40% might "not be important"; 30% to 60% may represent "moderate" heterogeneity; 50% to 90% may represent "substantial" heterogeneity; and 75% to 100% represents "considerable" heterogeneity. As noted in the Cochrane Handbook for Systematic Reviews of Interventions, we will keep in mind that the importance of I² depends on the magnitude and direction of effects; and on the strength of evidence for heterogeneity.
For the Chi² test, a P value ≤ 0.10 indicates evidence of statistical heterogeneity.
We reported the presence of substantial heterogeneity and investigated possible causes by following the recommendations provided in Section 9.6 of the Cochrane Handbook for Systematic Reviews of Interventions.
Assessment of reporting biases
For outcomes when more than 10 studies were present, we examined funnel plots to assess publication bias (Egger 1997; Sterne 2011). We used this approach to assess frequency of attacks and severity of attacks.
Data synthesis
We undertook meta‐analyses only when this was meaningful (i.e., when treatments, participants, and the underlying clinical question were similar enough for pooling to make sense). We used the generic inverse variance method for the meta‐analysis by entering the estimate of treatment effect and the standard error for each study. For RTSI 2000, we reported a geometric mean and found that the P value for attack frequency was not given with enough precision for calculation of a standard error for inclusion in the meta‐analysis.
We used a fixed‐effect model and performed a sensitivity analysis using the random‐effects model.
Subgroup analysis and investigation of heterogeneity
In addition to examining all CCBs versus placebo for any subset of RP, we performed the following subgroup analyses when possible.
-
Dihydropyridine type CCBs versus placebo. Dihydropyridine CCBs were studied as they are the subgroup of CCBs that are not cardioselective and are traditionally used in RP treatment whereas other CCBs such as verapamil are not routinely used and diltiazem is not used as first line subtype of CCBs.
-
CCBs versus placebo by dose (i.e., low dose, medium, medium/high dose). Low, medium and high doses for CCBs were defined as specified in the guideline in Appendix 2. Since only three studies used high doses of CCBs, we combined trials using high doses CCBs with the medium dose CCBs for outcomes where data for analysis was available. We had most data with respect to dose for nifedipine. Clinical impression is to start with lower doses and that if tolerated, higher doses may yield more benefit.
-
CCBs versus placebo by actual CCB drug (mainly nifedipine, nicardipine, and nisoldipine, as these CCBs were used most frequently in included trials).
-
CCBs versus placebo by Raynaud's type (primary or secondary).
-
Nifedipine versus placebo by disease type (primary or secondary RP).
For major outcomes with significant heterogeneity (I² > 50%), we tested robustness of results derived from the fixed effect by repeating the analysis using a random‐effects model. Because results from the two models were similar (i.e., in direction of effect), we reported fixed‐effect results throughout.
Sensitivity analysis
To address the presence of significant heterogeneity (I² > 50%; Higgins 2003), we performed post hoc sensitivity analyses. We repeated analyses of major outcomes with significant heterogeneity by omitting the study or studies believed to be responsible for the heterogeneity. Reasons for omission included the following.
-
Study used a scale of measure different from that used in other studies.
-
Study used study duration different from that used in other studies.
Grading the evidence
In addition to providing tools that can be used to assess risk of study bias, we used the GRADE approach in evaluating the overall quality of evidence for reported outcomes (Grade 2008). Through this approach, we assessed the quality of evidence as follows: (1) high quality from RCTs, (2) moderate quality downgraded by one level owing to a study limitation, (3) low quality double‐downgraded for study limitations, and, last, (4) very low quality triple‐downgraded owing to multiple study limitations. We downgraded evidence using the following GRADE approach.
-
Limitations in the design and implementation of available studies, suggesting high likelihood of bias.
-
Indirectness of evidence (indirect population, intervention, control, outcomes) (not applicable to this meta‐analysis, as only RCTs were analyzed).
-
Unexplained heterogeneity or inconsistency of results (including problems with subgroup analyses).
-
Imprecision of results (wide confidence intervals).
-
High probability of publication bias.
"Summary of findings" table
We have summarized the major outcomes of this review ‐ frequency of attack, duration of attack, severity of attack, pain, patient global assessment, withdrawals due to adverse events, and serious adverse events ‐ in summary of findings Table for the main comparison. This table contains intervention effect estimates, comparators, and details on quality of evidence.
Results
Description of studies
Refer to Characteristics of included studies,Characteristics of excluded studies, and Characteristics of studies awaiting classification for descriptions of individual studies.
Results of the search
The search performed on December 2, 2015, and updated on May 19, 2017, yielded 3389 hits (1061 from the Cochrane Library, 1027 from Medline, 1251 from Embase, and 50 from clinicaltrials.gov). After we screened out duplicates, 2337 articles remained. Further screening of these publications yielded 305 articles. When we examined the 305 articles further, we identified 77 articles, from which we excluded 30 with reasons and found that seven trials were awaiting classification, two trials were ongoing, and 38 trials met the inclusion criteria of this review. Figure 1 provides further details on our research results in a flow diagram format.
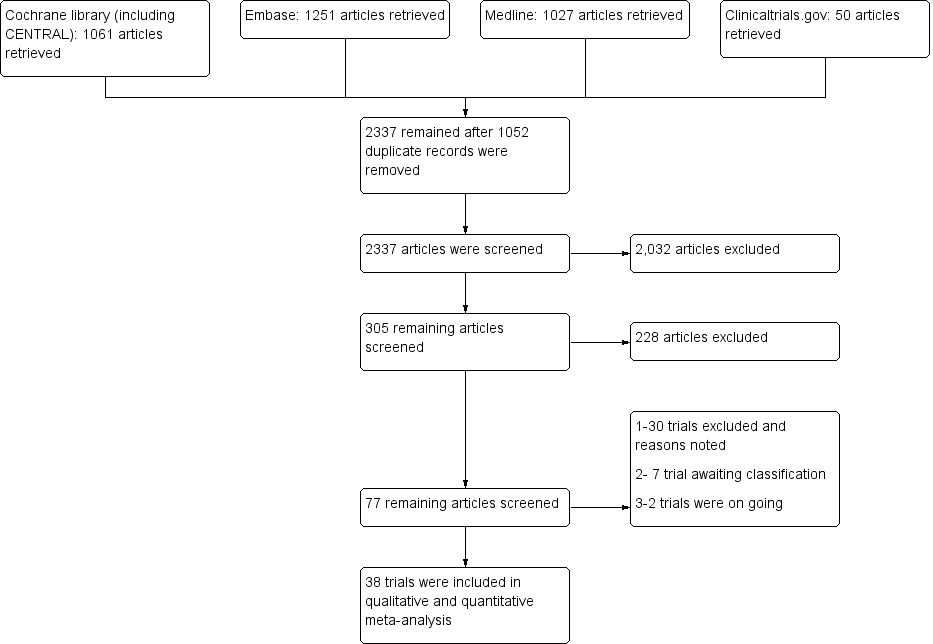
Flow diagram of study.
Included studies
We included 38 trials that investigated effects of CCBs versus placebo in 982 participants with RP (Aldoori 1986; Bravard 1983; Challenor 1987; Challenor 1989; Constantini 1987; Corbin 1986; Ettinger 1984; Ferri 1992; Finch 1988; French Co‐op 1991; Gjorup 1986a; Gjorup 1986b; Hawkins 1985; Kahan 1985a; Kahan 1985b; Kahan 1985c; Kahan 1987; Kallenberg 1987; Kinney 1982; Kirch 1987; La Civita 1997; Leppert 1989; Malamet 1984; Meyrick Thomas 1987; Muller‐Buhl 1983; Nilsson 1987; Rhedda 1985; Rodeheffer 1983; RTSI 2000; Rupp 1987; Sarkozi 1986; Sauza 1984; Smith 1982; Teixeira da Costa; Waller 1986; White 1986; Wigley 1987; Wollersheim 1991). See Characteristics of included studies.
Only nine of these studies exclusively studied patients with primary RP (N = 365 participants), five studied patients with secondary RP (N = 63 participants), and remaining studies (N = 554 participants) examined a mixture of patients with primary RP and secondary RP.
Of the 38 trials included in this systematic review, 36 were double‐blind and two were single‐blind; 33 were of cross‐over design and five were of parallel design. All studies were randomized, or appeared to be randomized. The duration of studies ranged from 2 to 20 weeks, with an average of 7.4 weeks and a median of 3 weeks per arm, with publication years ranging from 1982 to 2000.
Of the 38 included studies, 21 used low‐dose CCBs, 13 used medium‐dose CCBs, and three used high‐dose CCBs (see Appendix 2 for dosage ranges). Twenty‐two RCTs compared nifedipine versus placebo (Aldoori 1986; Bravard 1983; Challenor 1989; Constantini 1987; Corbin 1986; Finch 1988; Gjorup 1986b; Hawkins 1985; Kahan 1985a; Kahan 1985c; Kallenberg 1987; Kirch 1987; Malamet 1984; Meyrick Thomas 1987; Nilsson 1987; Rodeheffer 1983; RTSI 2000; Sarkozi 1986; Sauza 1984; Smith 1982; Waller 1986; White 1986). The daily dose of nifedipine ranged from 10 to 80 mg/d, the mean dose was 45 mg/d, and the median dose was 40 mg/d. Six trials compared nicardipine versus placebo (Ferri 1992; French Co‐op 1991; Kahan 1987; Rupp 1987; Wigley 1987; Wollersheim 1991a). Daily doses of nicardipine ranged from 30 to 100 mg, with a mean of 68 mg/d. Two trials compared nisoldipine versus placebo (Challenor 1987; Gjorup 1986a), providing daily dosages of 10 mg and 20 mg, respectively. Three trials compared diltiazem versus placebo (Kahan 1985b; Rhedda 1985; Teixeira da Costa), giving a mean dosage of 240 mg/d (dosages ranged from 180 mg to 360 mg, with a median dose of 180 mg/d). Individual studies compared Bay K 9320, amlodipine, isradipine, or verapamil (Muller‐Buhl 1983; La Civita 1997; Leppert 1989; Kinney 1982, respectively) versus placebo.
Excluded studies
We excluded 30 additional articles (see Characteristics of excluded studies). We excluded 10 studies because they lacked a placebo (Della Bella 1997; Dziadzio 1999; Leppert 1993; Myrdal 1994; Park 2013; Rademaker 1989; Rademaker 1992; Ringqvist 1993; Varela‐Aguilar 1997Wu 2008); another four studies because they did not present placebo data (Codella 1989; La Civita 1996; Rademaker 1989; Varela‐Aguilar 1997); nine trials because they were not randomized (Creager 1984; Garcia Hernandez2004; Joseph 1988; Kallenberg 1991; Lewis 1987; Pisenti 1984; Smith 1985; Vayssairat 1989; Wollersheim 1987); one trial because researchers gave participants placebo and treatment simultaneously (Schmidt 1989); two studies because they were not of adequate duration (duration < 1 week: Weber 1990; Kahan 1983b); one study because study authors reported an insufficient washout duration of one day (Winston 1983); and three were excluded because they were meta‐analyzes (Ennis 2016; Thompson 2001; Thompson 2005).
We have not currently included seven studies because we were unable to locate the articles or full data (EUCTR2009‐018194‐31‐GB; Kahan 1982; Kahan 1983a; Redondo 1986; van Heereveld 1988; Wasir 1983; Wise 1987). See Studies awaiting classification.
As of July 2017, we found two studies in Clinicaltrials.gov that were ongoing: One study compared 10% nifedipine versus 5% sildenafil (and placebo) (Vera‐Kellet 2017); the other compared diltiazem versus nitroglycerin and placebo (Nazarinia 2016).
Risk of bias in included studies
The most commonly encountered biases were lack of random sequence generation (in 78% of studies) and concealment of allocation (in 70% of studies), followed by performance, attrition, and selective reporting biases, respectively. See Characteristics of included studies, Figure 2, and Figure 3 for additional details on risk of bias of included studies.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
We judged allocation sequence generation as "high risk" for Rhedda 1985. See Characteristics of included studies. Although an independent collaborator performed randomization by using computer‐generated random numbers, randomization did not produce groups with equal baseline characteristics. At completion of the trial, researchers reported that the control group had more severe RP than the active treatment group. Nine of the 38 included studies adequately described allocation sequence and concealment, and we rated them as having "low risk" (Corbin 1986; French Co‐op 1991; Kahan 1985a; Kahan 1987; Nilsson 1987; Rhedda 1985; Rodeheffer 1983; RTSI 2000; Sarkozi 1986). Methods of concealment used by these trials included automated computer generation, tables of random numbers, block randomization, and randomization by a third party co‐ordinating center. The remainder of these studies failed to adequately describe how the allocation sequence and concealment were generated; we rated them as having "unclear risk of bias."
Blinding
Investigators stated that most (34/38; 89%) of the included RCTs were "double‐blind"; we assumed that these RCTs had adequate blinding and classified them as "low risk." Four studies (Hawkins 1985; Kirch 1987; Leppert 1989; Rodeheffer 1983) were "single‐blind," at least for some part of the trial. Therefore, we rated performance bias for these trials as showing "unclear risk", as seen in Figure 3.
With regard to blinding of outcome assessments, in some studies participants kept diaries for assessment of outcomes of interest (frequency, severity and duration of RP). Some studies used patient global assessments. Outcomes assessments appeared to be adequately blinded as all studies were blinded and 89% were double blind.
Incomplete outcome data
We found incomplete outcome data due to attrition in approximately 40% of the included trials. We assessed risk of bias due to incomplete outcome data as "unclear" in eight studies (Aldoori 1986; Challenor 1987; Constantini 1987; French Co‐op 1991; Muller‐Buhl 1983; RTSI 2000; Sauza 1984; Teixeira da Costa). These studies reported attrition with unclear effects on outcomes (i.e., it was clear whether attrition occurred in treatment or placebo phase in cross‐over studies, losses from groups were equal in small trials, dropout was uneven in larger trials). We classified eight trials as having "high risk" of bias with regard to attrition (Bravard 1983; Constantini 1987; Gjorup 1986b; Hawkins 1985; Rhedda 1985; RTSI 2000; Sarkozi 1986; Waller 1986). These trials reported high dropout rates, unequal dropout between treatment and placebo groups, and exclusion from analyses (due to missing data from subjective diary assessments or non‐compliance of participants). We classified the remaining 20 trials as having "low risk" of bias due to attrition; they had minimal or equal loss to follow‐up and treated participants on an intention‐to‐treat (ITT) basis.
Selective reporting
We rated selective reporting bias as "low risk" for most trials. Two trials did not report all proposed outcomes (Hawkins 1985; RTSI 2000). Hawkins 1985 noted that the severity of attacks on a 5‐point Likert scale was parallel to that on a 10‐cm VAS scale but did not report actual results; RTSI 2000 did not report all minor outcomes mentioned in the methods. We classified risk of reporting bias as "unclear" for these two trials.
Other potential sources of bias
Other potential risks of bias considered were possible carryover effects in cross‐over trials, and similarity of baseline characteristics in parallel trials. Overall, we judged these sources of bias as "low risk" for 18 trials (Aldoori 1986; Corbin 1986; Ferri 1992; Finch 1988; French Co‐op 1991; Kahan 1985a; Kirch 1987; La Civita 1997; Malamet 1984; Meyrick Thomas 1987; Muller‐Buhl 1983; Nilsson 1987; Rhedda 1985; Rodeheffer 1983; RTSI 2000; Sarkozi 1986; Sauza 1984; Teixeira da Costa). See Characteristics of included studies. For remaining included studies, we judged risk of bias for this domain as "unclear" owing to lack of sufficient information. Carryover bias was determined to be present if: there was no washout between treatments in a crossover trial (inadequate washout) and/or the baseline RP characteristics were dissimilar at the second treatment or the baseline characteristics for RP were not provided. CCBs are fast in their onset and rapidly washout when discontinued so the washout did not have to be for very long (such as one to two weeks) for patients to be assumed to be in steady state.
Effects of interventions
Primary outcomes
Comparison 1. All calcium channel blockers (CCBs) versus placebo for all subsets of Raynaud's phenomenon (RP)
See summary of findings Table for the main comparison.
Frequency of attacks
Average number of attacks/week in 528 participants from 23 studies
See Aldoori 1986; Challenor 1987; Challenor 1989; Corbin 1986; Ettinger 1984; Ferri 1992; Finch 1988; French Co‐op 1991; Gjorup 1986a; Hawkins 1985; Kahan 1985a; Kahan 1985b; Kahan 1985c; Kahan 1987; Kirch 1987; Malamet 1984; Meyrick Thomas 1987; Rodeheffer 1983; Rupp 1987; Sarkozi 1986; Smith 1982; Waller 1986; and Wigley 1987 (Analysis 1.1).
When we considered all trials regardless of the class or type of CCB, dose, or RP type, we found that the weighted mean difference (WMD) for the frequency of attacks per week was ‐6.07 (95% confidence interval [CI] ‐ 6.53 to ‐5.61). The negative sign in the WMD indicates that the intervention (CCBs) reduced the number of Raynaud's attacks per week when compared with placebo. This was equivalent to a relative reduction of 44% (95% CI ‐48% to ‐41%) in the frequency of attacks per week with CCBs. However, significant statistical heterogeneity (I² = 98%) was present. Excluding Kahan 1985c ‐ a short trial of two weeks per arm that showed a very large effect for CCBs compared with the remaining trials ‐ reduced heterogeneity to I² = 77. As a result, the new WMD was ‐2.93 (95% CI ‐3.44 to ‐2.43) (Analysis 1.2).
We did not include in the analysis RTSI 2000, which used a different scale of measure (geometric mean), provided an imprecise P value, and was much larger and longer than the other trials. This parallel trial compared effects of nifedipine versus placebo (77 participants took nifedipine, and 81 took placebo) in participants with primary RP and found a 66% reduction in frequency of attacks with nifedipine (P < 0.001) after one year. Additionally, participants taking nifedipine reported greater improvement in their RP symptoms compared with participants taking placebo (P < 0.001). Overall, effects of nifedipine on the frequency of attacks reported in this trial were consistent with the overall conclusion of this meta‐analysis.
Examination of the funnel plot of the frequency of attacks (Figure 4) showed an asymmetrical distribution of treatment effects around the mean estimate of effect (Egger 1997), with more studies showing benefit for CCBs.
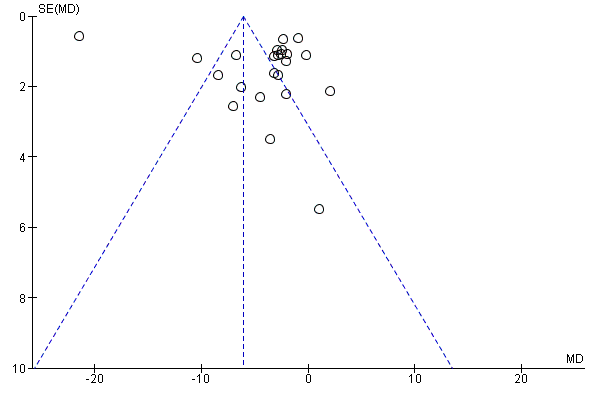
Funnel plot of comparison: 1 CCBs vs placebo (generic inverse variance method), outcome: 1.1 Frequency of attacks (average/week).
Duration of attacks
Average duration per attack measured in minutes in 69 participants from six studies
See Aldoori 1986; Ettinger 1984; Meyrick Thomas 1987; Finch 1988; Kirch 1987; and Malamet 1984 (Analysis 1.3).
The WMD for the average duration of attacks in the six trials was ‐1.67 minutes (95% CI ‐3.29 to 0) with considerable heterogeneity (I² = 89). This was equal to a small relative percent reduction of 9% (95% CI ‐18% to 0%).
Severity of attacks
Average severity per attack assessed on a 10‐cm visual analogue scale (0 = no symptoms, 10 = maximal severity) in 374 participants from 16 studies
See Challenor 1989; Ettinger 1984; Ferri 1992; Finch 1988; French Co‐op 1991; Gjorup 1986a; Hawkins 1985; Kahan 1985a; Kahan 1985b; Kahan 1985c; Kahan 1987; Kirch 1987; Malamet 1984; Rupp 1987; Smith 1982; and Wigley 1987 (Analysis 1.4).
The pooled WMD was ‐0.62 cm (95% CI ‐0.72 to ‐ 0.51) with significant heterogeneity (I² = 92%). The negative sign in the WMD indicates that the severity of attacks was less among the active treatment group than the placebo group. Absolute risk difference was ‐6% (95% CI ‐11% to ‐8%) and relative percent change was ‐9% (95% CI ‐11% to ‐8%).
Examination of the funnel plot (Figure 5) for severity of attacks showed some asymmetry with a few outliers in the direction of effect.
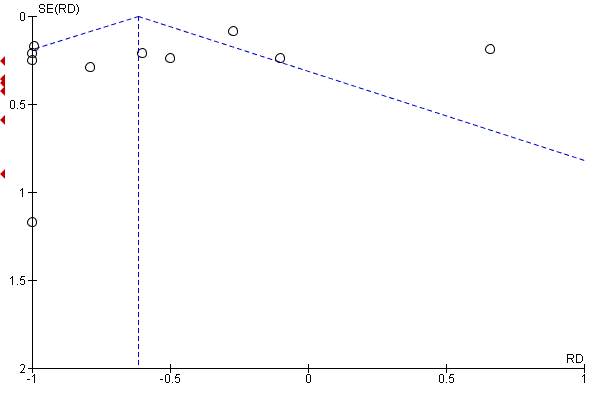
Funnel plot of comparison: 11 CCBs vs placebo (generic inverse variance method), outcome: 11.4 Severity of attacks (average, on a 10‐cm VAS).
Pain
Average pain per attack measured on a 10‐cm visual analogue scale (0 = no pain, 10 = maximal pain) in 62 participants from 4 studies
See Aldoori 1986; Ettinger 1984; Malamet 1984; and Rupp 1987 (Analysis 1.5).
The pooled summary WMD for pain in these studies was ‐1.47 cm (95% CI ‐2.21 to ‐0.74). The negative sign indicates that pain was less in the active treatment group. Absolute risk reduction was ‐15% (95% CI ‐22 % to ‐7%) and relative percent change was ‐47% (95% CI ‐71% to ‐24%). Heterogeneity was moderate (I² = 77%).
Patient global
Disability due to Raynaud's assessed on a 10‐cm visual analogue scale (0 = no disability, 10 = maximal disability) in 96 participants from two studies
See Challenor 1987 and French Co‐op 1991 (Analysis 1.6).
The WMD for patient global was ‐0.37 (95% CI ‐0.73 to 0). We did not calculate the number needed to treat for an additional beneficial outcome (NNTB) for this non‐significant result, but absolute risk difference was ‐4% (95% CI ‐7% to 0%) and relative percent change was ‐9% (95% CI ‐19% to 0%).
Withdrawals
Numbers of participants who dropped out of studies owing to adverse treatment effects among a total of 63 participants from two studies
See Constantini 1987 and Sarkozi 1986 (Analysis 1.7).
The pooled summary risk ratio (RR) of withdrawals from two parallel trials that reported this outcome was 1.32 (95% CI 0.51 to 3.33); 8 out of 32 withdrew from active treatment compared with 6 out of 31 from placebo treatment. Absolute risk difference was 6% (95% CI ‐14% to 26%) and relative percent change was 30% (95% CI ‐49% to 233%). We did not calculate the number needed to treat for an additional harmful outcome (NNTH) because withdrawals did not differ statistically between the two groups.
We did not analyze withdrawals for cross‐over trials but did notice that overall withdrawals were more frequent with CCBs than with placebo. From 10 cross‐over trials with 281 participants that reported withdrawals, 39 participants withdrew while on active treatment compared with 15 while taking placebo (see Appendix 3).
Serious adverse events
Number of participants who died or withdrew and were hospitalized as a result of adverse effects of treatment
Investigators reported no serious adverse events
Note: Wollersheim 1991 met the inclusion criteria of this review, but the only outcome that could be included in the meta‐analysis was withdrawals. For the other reported outcomes ‐ frequency, duration, and severity of attacks ‐ trial authors reported no statistically significant differences between groups but provided no estimates of variance. This trial used medium‐dose nicardipine and included a mixture of participants with primary and secondary RP. If we had been able to include in our meta‐analysis trial data on frequency, duration, and severity of attacks, we expect that the non‐significant results would decrease observed effects of CCBs on these outcomes. Also, we did not include RTSI 2000 in the meta‐analysis, as this trial reported geometric mean differences and did not report the P value with enough precision for calculation of a standard error.
Subgroup analyses included the following comparisons.
Comparison 2. CCBs versus placebo by RP type (primary vs secondary)
Frequency of attacks
Average number of attacks/week in 528 participants from 12 studies
See Challenor 1989; Corbin 1986; Ettinger 1984; Kahan 1985a; Kahan 1985b; Kahan 1987; Kirch 1987; Malamet 1984; Meyrick Thomas 1987; Rodeheffer 1983; Rupp 1987; and Sarkozi 1986 (Analysis 2.1).
In 226 people with primary RP, CCBs reduced the frequency of attacks per week by 3.02 (95% CI ‐3.65 to ‐2.38) compared with placebo. In 102 people with secondary RP, CCBs reduced the average number of attacks over a one‐week period by 3.42 (95% CI ‐4.33 to ‐2.51) compared with placebo. These differences between groups were not statistically significant (P = 0.48; I² = 48%).
Severity of attacks
Average severity per attack assessed on a 10‐cm visual analogue scale (0 = no symptoms, 10 = maximal severity) in 253 participants from 10 studies
See Challenor 1989; French Co‐op 1991; Gjorup 1986a; Hawkins 1985; Kahan 1985a; Kahan 1985b; Kahan 1987; Kirch 1987; Malamet 1984; and Rupp 1987 (Analysis 2.2).
Compared with placebo, CCBs reduced the severity of attacks by 0.95 on a 10‐cm scale (95% CI ‐1.11 to ‐0.79; I² = 96%) in 184 people with primary Raynaud's. In 69 people with secondary RP, CCBs reduced the severity of attacks by 0.48 (95% CI ‐0.61 to ‐0.35; I² = 94%) compared with placebo.
The difference between these subgroups was statistically significant (P < 0.0001; I² = 95%).
Comparison 3. Nifedipine versus placebo by RP type (primary vs secondary)
Frequency of attacks
Average number of attacks/week in 233 participants from nine studies
See Challenor 1989; Corbin 1986; Ettinger 1984; Kahan 1985a; Kirch 1987; Malamet 1984; Meyrick Thomas 1987; Rodeheffer 1983 and Sarkozi 1986 (Analysis 3.1).
Among 90 participants with primary RP, nifedipine reduced the frequency of attacks by 4.42 (95% CI ‐5.35 to ‐3.50) compared with placebo. In 60 participants with secondary RP, nifedipine reduced the frequency of attacks by 4.19 (95% CI ‐5.47 to ‐2.91) compared with placebo. However, heterogeneity was substantial (I² = 98% and I² = 87%, respectively).
The difference between these subgroups was not statistically significant (P = 0.77).
Severity of attacks
Average severity per attack assessed on a 10‐cm visual analogue scale (0 = no symptoms, 10 = maximal severity) in 39 participants from four trials
See Challenor 1989; Kahan 1985a; Kirch 1987; and Malamet 1984 (Analysis 3.2).
In 27 participants with primary RP, nifedipine when compared with placebo reduced the severity of attacks (WMD 1.74, 95% CI ‐2.09 to ‐1.39). In 27 participants with secondary RP, nifedipine did not appear to be any more beneficial than placebo (WMD 0.01, 95% CI ‐0.32 to 0.34). However, heterogeneity was substantial (I² = 98% and I² = 96%, respectively).
The difference between these subgroups was statistically significant (P < 0.0001; I² = 97%).
Comparison 4. CCBs versus placebo for primary and secondary RP by CCB class (dihydropyridine class vs non‐dihydropyridine class)
For frequency and severity of attacks, we had enough data to perform a subgroup analysis by CCB class (dihydropyridine vs non‐dihydropyridine). We further analyzed the data for these outcomes by type of dihydropyridine CCB (i.e., nifedipine vs placebo, nicardipine vs placebo, and nisoldipine vs placebo).
Frequency of attacks (average/week)
Average number of attacks/week in 528 participants from 23 studies
See Aldoori 1986; Challenor 1987; Challenor 1989; Corbin 1986; Ettinger 1984; Ferri 1992; Finch 1988; French Co‐op 1991; Gjorup 1986a; Hawkins 1985; Kahan 1985a; Kahan 1985b; Kahan 1985c; Kahan 1987; Kirch 1987; Malamet 1984; Meyrick Thomas 1987; Rodeheffer 1983; Rupp 1987; Sarkozi 1986; Smith 1982; Waller 1986; and Wigley 1987 (Analysis 4.1).
Of the 23 studies included in this analysis, 22 used the dihydropyridine class of CCBs, and only one used the non‐dihydropyridine class (Kahan 1985b). The WMD for frequency of attacks in the dihydropyridine class of CCBs compared with placebo was ‐6.13 (95% CI ‐6.60 to ‐5.67).
We further analyzed the data for frequency of attacks by dihydropyridine CCB type. Among 22 studies using the dihydropyridine class, 15 used nifedipine, 4 nicardipine, and 2 nisoldipine. In trials comparing nifedipine versus placebo, nifedipine reduced the number of attacks by 8.62 (95% CI ‐9.20 to ‐8.03; I² = 98%) in 290 people; nicardipine reduced the frequency of attacks by 1.92 (95% CI ‐2.80 to ‐1.04; I² = 50%) in 150 people; and nisoldipine reduced the frequency of attacks by 3.00 per week (95% CI ‐4.57 to ‐1.43; I² = 0%) in 39 people (Analysis 4.2).
Severity of attacks
Average severity per attack assessed on a 10‐cm visual analogue scale (0 = no symptoms, 10 = maximal severity) in 374 participants from 16 studies
See Challenor 1989; Ettinger 1984; Ferri 1992; Finch 1988; French Co‐op 1991; Gjorup 1986a; Hawkins 1985; Kahan 1985a; Kahan 1985b; Kahan 1985c; Kahan 1987; Kirch 1987; Malamet 1984; Rupp 1987; Smith 1982; and Wigley 1987 (Analysis 4.3).
Only 1 of the 16 trials providing data on severity used the non‐dihydropyridine class, and 15 used the dihydropyridine class. The WMD for severity of attacks in the 15 trials comparing dihydropyridine CCBs versus placebo was ‐0.60 (95% CI ‐0.71 to ‐0.50). Heterogeneity was substantial (I² = 92%).
Further analysis of data by dihydropyridine CCB type shows that when compared with placebo, nifedipine reduced the severity of attacks by 0.79 (95% CI ‐0.96 to ‐0.61; I² = 94%) in 189 people; nicardipine reduced the severity of attacks by 0.47 (95% CI ‐0.61 to ‐0.33; I² = 91%) in 150 people; and nisoldipine reduced the severity of attacks by 0.79 (95% CI ‐1.36 to ‐0.22) in 19 people (Analysis 4.4).
Comparison 5. CCBs versus placebo in comparison by CCB dose for RP (both primary and secondary RP included)
Most of the included trials used low‐dose CCBs (refer to Appendix 2 for dosage ranges), and few used high‐dose CCBs. Hence, for this subgroup analysis, we examined low‐dose CCBs versus placebo, and medium/high‐dose CCBs versus placebo.
Frequency of attacks
Average number of attacks/week in 528 participants from 23 studies
See Aldoori 1986; Challenor 1987; Challenor 1989; Corbin 1986; Ettinger 1984; Ferri 1992; Finch 1988; French Co‐op 1991; Gjorup 1986a; Hawkins 1985; Kahan 1985a; Kahan 1985b; Kahan 1985c; Kahan 1987; Kirch 1987; Malamet 1984; Meyrick Thomas 1987; Rodeheffer 1983; Rupp 1987; Sarkozi 1986; Smith 1982; Waller 1986; and Wigley 1987 (Analysis 5.1).
When we examined the frequency of attacks by CCB dose versus placebo, we found that the pooled WMD for frequency of attacks with low‐dose CCBs was ‐3.00 (95% CI ‐3.63 to ‐2.37; I² = 68%) compared with ‐9.50 (95% CI ‐10.17 to ‐8.83; I² = 92%) with medium/high‐dose CCBs. However, medium/high‐dose CCBs showed substantial heterogeneity (I² = 99%). A sensitivity analysis of medium/high‐dose CCBs versus placebo that excluded Kahan 1985a and Kahan 1985c, whose treatment effect was much larger than that noted in the remaining studies, eliminated heterogeneity and revealed that the WMD for frequency of attacks between CCBs and placebo was ‐1.74 (95% CI ‐2.63 to ‐0.85; I² = 0%).
Differences between subgroups by dosage were statistically significant (P < 0.0001; I² = 98%).
Duration of attacks
Average duration per attack measured in minutes in 69 participants from six studies
See Aldoori 1986; Ettinger 1984; Finch 1988; Kirch 1987; Malamet 1984; and Meyrick Thomas 1987 (Analysis 5.2).
The pooled WMD for the duration of attacks with low‐dose CCBs was 2.24 (95% CI ‐0.24 to 4.73) compared with placebo for 56 participants. However, heterogeneity was substantial (I² = 92%). The WMD for the duration of attacks with medium‐dose CCBs was ‐4.60 (95% CI ‐6.76 to ‐2.45) compared with placebo for 82 participants.
Differences between these subgroups were statistically significant (P < 0.0001; I² = 94%).
Severity of attacks
Average severity per attack assessed on a 10‐cm visual analogue scale (0 = no symptoms, 10 = maximal severity) in 374 participants from 16 studies
See Challenor 1989; Ettinger 1984; Ferri 1992; Finch 1988; French Co‐op 1991; Gjorup 1986a; Hawkins 1985; Kahan 1985a; Kahan 1985b; Kahan 1985c; Kahan 1987; Kirch 1987; Malamet 1984; Rupp 1987; Smith 1982; and Wigley 1987 (Analysis 5.3).
The WMD for severity of attacks was ‐0.56 (95% CI ‐0.68 to ‐0.45; I² = 94%) when low‐dose CCBs were compared with placebo in 217 people, and ‐0.91 (95% CI ‐1.18 to ‐0.64; I² = 90%) when medium/high‐dose CCBs were compared with placebo in 157 people.
Differences between these subgroups were statistically significant (P = 0.02; I² = 81%).
Pain
Average pain per attack measured on a 10‐point visual analogue scale (0 = no pain, 10 = maximal pain) in 62 participants from four studies
See Aldoori 1986; Ettinger 1984; Malamet 1984; and Rupp 1987 (Analysis 5.4).
The WMD for pain was ‐3.04 (95% CI ‐4.34 to ‐1.75; I² = 59%) for 36 participants with low‐dose CCBs, and ‐0.73 (95% CI ‐1.62 to 0.16) for 26 participants with medium‐dose CCBs when each was compared with placebo.
Again, differences between these subgroups were statistically significant (P = 0.04; I² = 88%).
Patient global
Disability due to Raynaud's assessed on a 10‐cm visual analogue scale (0 = no disability, 10 = maximal disability) in 96 participants from two studies
See Challenor 1987 and French Co‐op 1991 (Analysis 5.5).
Challenor 1987 compared a low‐dose CCB versus placebo in 36 people and reported that the WMD for patient global was ‐0.20 (95% CI ‐0.63 to 0.23). French Co‐op 1991 compared a high‐dose CCB versus placebo in 60 people and reported that the WMD for patient global was ‐0.74 (95% CI ‐1.37 to ‐0.11). Differences between these two trials were not statistically significant (P = 0.16; I² = 48%).
Minor outcomes
See Analysis 6.1 to Analysis 6.2 (Appendix 3).
For our minor outcomes, trials reported general improvement, treatment preference, changes in digital ulcers, and side effects.
General improvement
Three parallel RCTs reported on and analyzed general improvement (Constantini 1987; Sarkozi 1986; Sauza 1984). For these trials, the risk ratio of improvement was 2.38 (95% CI 1.35 to 4.20; I² = 12%). Data from cross‐over trials were not analyzed; however, more participants from these trials reported improvement while receiving active treatment over placebo (nine cross‐over trials with 109 participants reported 76 cases of general improvement on active treatment vs 29 cases on placebo).
Treatment preference
Four cross‐over trials with a total of 89 participants reported treatment preference (Corbin 1986; Gjorup 1986a; Gjorup 1986b; Rupp 1987). Of the these 89 participants, 61 preferred active treatment and 17 indicated a preference for placebo.
Changes in digital ulcers
Only one study considered changes in digital ulceration (Meyrick Thomas 1987). This study reported 18 new digital ulcers in six patients taking placebo and nine new ulcers among three patients taking nifedipine.
Side effects
Three parallel trials reported side effects and thus were meta‐analyzed for this outcome (Constantini 1987; Sarkozi 1986; Sauza 1984). The RR for side effects was 1.12 (95% CI 0.87 to 1.45). Hence, side effects were more common with CCBs than with placebo. This was also true for the cross‐over trials: Of 573 participants from 26 cross‐over trials, 265 experienced side effects on active treatment compared with 85 on placebo.
Results from the search of regulatory websites
On March 8, 2016, we searched for black box warnings on websites for FDA MedWatch, European Medicines Evaluation Agency, Australian Adverse Drug Reactions Bulletin, and UK Medicines and Healthcare products Regulatory agencies. We found no black box warnings for nifedipine, nicardipine, nisoldipine, diltiazem, verapamil, amlodipine, isradipine, or BAY K 9320.
Discussion
Summary of main results
For this review, we identified 38 studies with 982 participants and considered all subsets of Raynaud's phenomenon (RP) (both primary and secondary). However, not all studies reported all outcomes of interest. We used the Cochrane risk of bias assessment tool as well as the GRADE approach to evaluate the quality of evidence. In addition, we examined effects of calcium channel blockers (CCBs) by class, type, and dose, as well as by Raynaud's type (primary vs secondary). The most frequently encountered risk of bias types were incomplete outcome data and lack of reporting of methods used for randomization generation and allocation. For most outcomes, we downgraded evidence for small sample sizes because included trials did not always report all outcomes. Overall, evidence quality ranged from low to moderate (three moderate quality and three low quality).
Major outcomes
CCBs (all) versus placebo for all RP subsets
When we examined CCBs compared with placebo for all RP subsets, we found that CCBs (given for between 2 and 20 weeks) were superior in reducing the frequency, severity, and pain of attacks, as seen in summary of findings Table for the main comparison. Reductions in frequency and severity of Raynaud's attacks were small to moderate and were based on evidence of moderate quality derived from 23 trials with 528 participants, and from 18 trials with 415 participants, respectively. Reduced frequency of attacks with CCBs remained after a post hoc sensitivity analysis was performed to address some of the observed heterogeneity. Other plausible explanations for the remainder of the heterogeneity noted for these outcomes included variations in the duration of trials, in CCB dose and type, and in methodological rigor.
Evidence of low quality from four studies with 62 participants shows that CCBs were better than placebo for reducing pain associated with Raynaud's attacks. The impact of CCBs on the duration of attacks and on patient global, although favoring CCBs, was uncertain because evidence on small numbers of participants was obtained from a limited number of trials measuring and reporting these outcomes.
Two parallel studies with 69 participants analyzed withdrawals due to adverse events and found that withdrawals were more common with CCBs than with placebo; however, this evidence was of low quality and differences may not have been important. For cross‐over trials, we did not analyze withdrawals (because of the challenges involved in analyzing dichotomous outcomes from cross‐over trials), but we noted that overall, more withdrawals were reported with CCBs than with placebo. These studies did not report serious adverse events (leading to withdrawal followed by hospitalization or death) directly attributed to treatment.
CCBs versus placebo by RP type (primary vs secondary)
Examination of all CCBs versus placebo by RP type revealed that CCBs (when compared with placebo) had a similar effect in both subgroups with regard to reducing the frequency of attacks (a reduction of three attacks/week). However, the number of participants with primary RP was much larger than the number with secondary RP. Hence, this difference in sample sizes may have affected observed effect estimates. Another comparison revealed that CCBs were superior to placebo in primary RP with regard to reducing the severity of attacks (‐0.91 cm vs ‐0.48 cm, respectively). The smaller treatment effect sizes noted in secondary RP with regard to RP severity are consistent with those reported in clinical practice owing to the more severe nature of the disease in this subpopulation.
Nifedipine versus placebo by RP type (primary or secondary)
Comparison of nifedipine versus placebo by RP type showed that nifedipine was slightly better in reducing the frequency of attacks in primary RP (‐4.42 vs ‐4.19, respectively); and nifedipine was much more effective in reducing the severity of attacks in primary than in secondary RP (‐1.74 cm vs 0.01 cm, respectively). However, sample sizes for these subgroup analyses were very small, likely making analyses underpowered to detect the true effects of treatment. In general, improvement in secondary RP may be less than in primary RP, as the condition in the former group is likely more severe and potentially less reversible with fixed vascular changes ‐ not just vasospasm.
Subgroup analysis by CCB class: dihydropyridines (most common class of CCBs) versus placebo for all RP subsets
Comparison of only the dihydropyridine class of CCBs versus placebo for all RP subsets yielded similar results to those obtained when all CCBs were compared with placebo; this was expected because all trials but one used the dihydropyridine class of CCBs.
Nifedipine, nicardipine, and nisoldipine were analyzed separately versus placebo for frequency and severity of attacks. All three CCBs were superior to placebo in decreasing both the frequency and the severity of attacks. Nifedipine was used by most of the included trials and led to the largest reduction in frequency of attacks. Nifedipine also had a larger effect estimate with regard to severity of attacks when compared with nicardipine ‐ the second most commonly used CCB. Nisoldipine was used by only two trials and had a larger effect size on severity of attacks, but this effect estimate was likely imprecise owing to the small sample size. Overall, nifedipine appeared to be the most beneficial treatment with regard to reducing the frequency and severity of attacks.
CCBs (by dose) versus placebo for all RP subsets
Comparison of CCBs versus placebo by CCB dose (low, medium/high) revealed that higher doses were superior to lower doses in reducing the frequency, duration, and severity of attacks, as well as in improving patient global. In addition, although most trials used low‐dose CCBs, and fewer used medium to high doses, results show that higher doses had larger treatment effects and hence appeared more beneficial with regard to the frequency, duration, and severity of attacks, as well as improved patient global. Although significant heterogeneity was evident, low‐dose CCBs reduced the frequency of attacks per week by 3.0 and the severity of attacks by 0.56 cm (on a scale of 0 to 10 cm), and medium/high‐dose CCBs reduced the frequency of attacks by 9.5 and severity by 0.91 cm. In addition, medium/high‐dose CCBs were superior to placebo in reducing the average duration of attacks (mean difference [WMD] ‐4.60, 95% CI ‐6.76 to ‐2.45), but low‐dose CCBs compared with placebo did not produce this effect (WMD 2.24, 95% CI ‐0.24 to 4.73). Higher doses of CCBs were better in reducing disability due to RP (patient global): A study comparing a high‐dose CCB versus placebo in 60 participants found a reduction of 0.74 cm (95% CI ‐1.37 to ‐0.11) with CCBs, but another trial using a low‐dose CCB versus placebo in 36 participants found a reduction of 0.20 cm (95% CI ‐0.63 to 0.23).
Researchers found a larger reduction in pain with low‐dose CCBs versus placebo than with higher‐dose CCBs versus placebo. However, overall sample sizes were small, and a larger number of trial participants used low‐dose CCBs compared with placebo.
Overall, higher doses of CCBs compared with placebo appear to be more beneficial with regard to the primary outcomes examined. Larger treatment effect estimates of CCBs at higher doses are consistent with findings reported in clinical practice, but dosages generally are based on patient tolerability.
Heterogeneity and publication bias
We explored explanations for the presence of heterogeneity in treatment effects by performing subgroup and sensitivity analyses. Overall, we observed heterogeneity with most outcomes that was partially explained by differences in the type and dose of CCBs, as well as by differences in disease type (primary vs secondary RP) and study duration, with most trials being of very short duration. Other plausible factors that may have contributed to heterogeneity but were not explored include differences in inclusion criteria of trials, rigor in trial methods, trial type (cross‐over vs parallel), and differences in the time of year trials were performed. To our knowledge, only one study reported that concomitant medications were continued during the trial. Hence, we do not believe that use of concomitant vasodilator medications was a significant confounder in the observed results.
For major outcomes reported in at least 10 trials (frequency and severity of attacks), we examined funnel plots to assess for publication bias. Both funnel plots showed some asymmetry around the mean effect estimate but did not indicate publication bias as a major concern. Plausible explanations for the presence of asymmetry in these funnel plots include the presence of significant heterogeneity, differences in the quality and size of trials, differences in methods (i.e., parallel vs cross‐over design), and possibly selective reporting.
Minor outcomes
With regard to our minor outcomes, analysis of three parallel trials showed that CCBs were associated with more general improvement in patients' Raynaud's condition. We were unable to meta‐analyze dichotomous outcomes from cross‐over trials, but we did examine overall trends. Cross‐over trials also reported that more participants reported improvement while taking CCBs than placebo, and more participants preferred active treatment over placebo. One trial reporting changes in digital ulceration found that nine participants developed 18 new digital ulcers while taking placebo, and three participants developed six new digital ulcers while taking nifedipine. Results from this trial suggest that CCBs such as nifedipine may be useful for prevention of digital ulcers. However, participants who took CCBs reported more side effects than were reported by those who took placebo, but none of these reported adverse effects were serious.
Conclusions from this review and meta‐analysis
Overall, although they were associated with more non‐serious side effects, CCBs were preferred to placebo and were better in reducing the frequency, duration, and severity of Raynaud's attacks when given over 2 to 20 weeks. In addition, CCBs produced a potentially clinically important mean improvement in pain, and more participants taking CCBs reported general improvement. In particular, CCBs showed greater benefit at higher doses in reducing the frequency and severity of attacks for patients with primary RP. It is important to note that researchers reported no serious adverse events directly related to treatment with CCBs. These findings indicate that CCBs, particularly those in the dihydropyridine class (i.e., nifedipine and nicardipine), given at higher doses may be beneficial for the management of RP (especially among patients with primary RP) requiring pharmacological intervention (although most people with RP, especially primary RP, do not require pharmacological treatment). Some clinically important outcomes of interest (i.e., changes in average duration of attacks, pain, and patient global) were measured or reported by only a few trials. Hence, effect estimates for these outcomes were imprecise but appeared to favor CCBs. The findings of this review may be generalizable to other members of the dihydropyridine class of CCBs, such as amlodipine, bepridil, felodipine, isradipine, and nisoldipine.
Overall completeness and applicability of evidence
Most of the trials included in this review were small and included a mixture of patients with primary and secondary RP. Fewer trials enrolled patients with secondary RP, which makes it more difficult to generalize the results of this review to these patients. Review results indicate that CCBs (particularly those in the dihydropyridine class) may be beneficial in the management of RP, especially primary RP. Furthermore, both nifedipine and nicardipine ‐ the most commonly used dihydropyridines ‐ proved superior to placebo in reducing the frequency and severity of RP attacks. However, several concerns must be addressed to guide clinicians in using or interpreting the findings of this review.
-
The overall quality of evidence ranged from low to moderate, with evidence for three outcomes being of low quality and three of moderate quality. Many trials included in the meta‐analyses were not methodologically rigorous. This variation in the quality of the evidence requires cautious interpretation and use of review findings, especially in clinical practice.
-
Most trials had small sample sizes. As a result, we were unable to conduct subgroup analysis for all major outcomes of this review. The overall small sample size was a problem, especially for some outcomes such as duration and patient global, which were not measured or reported by many trials. For outcomes with small sample sizes, results often have large confidence intervals and are imprecise.
-
Most included trials were of the cross‐over design. Potential carryover effects between active treatment and placebo are possible with this method. We were unable to estimate a period effect. Results may be biased.
-
The dosage of CCBs used was highly variable, with most trials using low‐dose CCBs. We found evidence of higher efficacy for medium/high versus.low doses of CCBs in RP treatment. Hence, suboptimal doses in most included trials may be responsible for the non‐significant findings observed for some outcomes. Clinicians may start with low doses of CCBs and may increase dosage to achieve a desired effect (if not obtained with low dose), as long as the patient can tolerate it and no serious side effects develop.
-
Most included trials include a mixture of patients with primary and secondary RP. Only seven trials exclusively included patients with primary RP, and even fewer included only those with secondary RP. Consistent with the findings of this study, patients with primary RP often respond better to medication, possibly as a result of the milder nature of their disease. Only a few studies including patients with secondary RP reported the important outcomes of interest. Most trials did not stratify randomization between primary and secondary RP (or within secondary RP between systemic sclerosis, which is the most severe form of secondary RP, and other connective tissue disorders). This could have introduced bias, as groups may not have been equal with respect to the proportion of primary and secondary RP between treatment arms (for parallel designed studies).
-
Another problem could be the variability of treatment duration between trials. However, CCBs are fast acting, and most patients respond to treatment quickly. Thus efficacy of CCBs would be expected in short trials, but longer trials may report increased dropout and greater regression to the mean and/or attenuation of benefit for other reasons.
-
Included trials showed high variability in outcome measurements and scales used. To allow for ease of interpretation, we converted all results to the most commonly used scales. For instance, for severity of attacks, we converted the results of all trials to a 10‐cm visual analogue scale score. This variability in the scales of measure likely contributed to some of the observed heterogeneity.
-
Last, significant statistical heterogeneity was seen with most outcomes. Some heterogeneity was partially reduced by subgroup and sensitivity analyses. However, heterogeneity often remained and is likely a result of the different factors discussed above.
Quality of the evidence
We assessed the quality of evidence from trials included in this review using the GRADE approach (Grade 2008). Many trials, particularly older ones from the 1980s (when reporting was less standardized and protocols were not registered) failed to fully describe their methods. The deficiency that we most commonly encountered when assessing risk of bias in these trials was lack of full descriptions of random sequence generation and allocation concealment. Most publications noted that the study was "randomized" and "double‐blind" without providing further details.
We downgraded outcomes of frequency of attacks and severity of attacks to moderate quality owing to concerns about inconsistency of results across trials. We downgraded evidence on patient global assessment to moderate quality for concerns about imprecision due to the small sample size. We downgraded evidence on duration of attacks and pain to low quality for concerns about both inconsistency and imprecision. We downgraded evidence quality to low for withdrawals due to adverse events, given the small sample size (imprecision) and high attrition rate noted in the two studies that reported this outcome.
Given the moderate to low quality of evidence on major outcomes, one should use caution, but trial results do mirror what is seen in clinical practice; hence they have face validity despite methodological flaws.
Potential biases in the review process
We performed an extensive and sensitive electronic search of all important databases to ensure retrieval of as many studies as possible. In addition, at least two review authors performed most of the steps of the review and consulted with a third review author regarding disagreements.
Some subgroup analyses were post hoc which may have introduced bias as many trials did not stratify randomization on the analyses of interest. We did plan to study: primary vs. secondary RP and RP from systemic sclerosis (as the scleroderma patients are more difficult to treat and have more severe and complicated RP), we planned to study subsets of CCBs (dihydropyridines and nifedipine in particular as we knew the bulk of the data was from nifedipine), but studying some other subgroups post hoc may have been biased. Overall, we did not identify any major biases in the review process.
Agreements and disagreements with other studies or reviews
The findings of this review, although broader in scope, are similar to those of our 2005 meta‐analysis ‐ Thompson 2005 ‐ and of a recent meta‐analysis ‐ Ennis 2016 ‐ on the use of oral vasodilators (including CCBs) for primary RP. Both of these meta‐analyses noted reduced frequency of attacks among patients with primary RP when treated with CCBs. Thompson 2005 found that CCBs decreased the frequency of attacks in patients with primary RP by a mean of 2.8 to 5 attacks per week, and Ennis 2016 noted a decrease of 0.6 to 2.8 attacks per week. In the current review, when review authors considered both primary and secondary RP, we found an average decrease in the frequency of six attacks per week (and an average decrease of three attacks per week with exclusion of Kahan 1985c). When considering only primary RP, we found an average decrease of three attacks per week. This difference between our study and Ennis 2016 may be due to the fact that Ennis 2016 included all trials examining CCBs versus placebo or other treatments. We limited our review to trials examining only CCBs versus placebo and no other oral vasodilators from other classes of medications.
The Thompson 2001 meta‐analysis shows that CCBs significantly reduced both the frequency and the severity of attacks in patients with systemic sclerosis. This review did not find that CCBs significantly reduced any of the reported major outcomes in patients with secondary RP. Differences between these reviews are likely due to the fact that review authors for Thompson 2001 included a larger number of trials (any trials with over 75% of trial participants with systemic sclerosis) and performed subset analyses of data from trials that included participants with systemic sclerosis, if data could be extracted. In our current review of secondary RP, we included only trials in which all participants had secondary RP of any origin, resulting in very small numbers of trials and participants. We included any trial of participants with secondary RP in which nearly all had connective tissue disease.
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Funnel plot of comparison: 1 CCBs vs placebo (generic inverse variance method), outcome: 1.1 Frequency of attacks (average/week).
Funnel plot of comparison: 11 CCBs vs placebo (generic inverse variance method), outcome: 11.4 Severity of attacks (average, on a 10‐cm VAS).

Comparison 1 CCBs vs placebo (generic inverse variance method), Outcome 1 Frequency of attacks (average/week).

Comparison 1 CCBs vs placebo (generic inverse variance method), Outcome 2 Frequency of attacks (average/week).

Comparison 1 CCBs vs placebo (generic inverse variance method), Outcome 3 Duration of attacks (minutes).
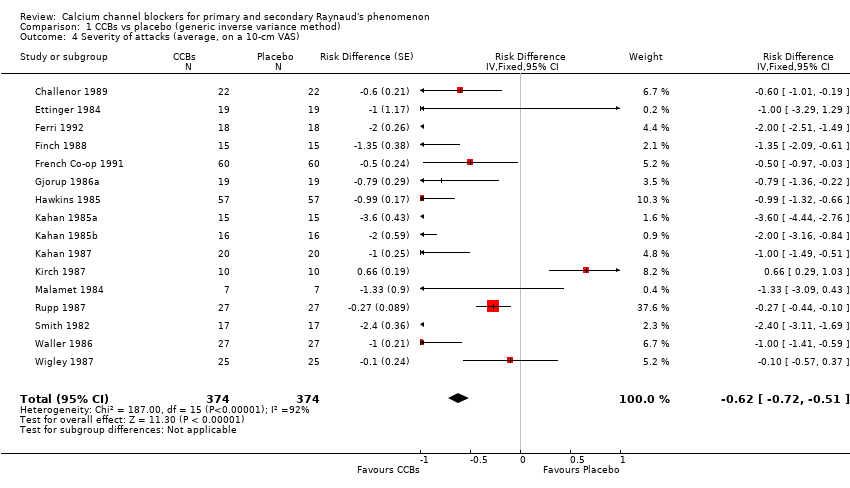
Comparison 1 CCBs vs placebo (generic inverse variance method), Outcome 4 Severity of attacks (average, on a 10‐cm VAS).

Comparison 1 CCBs vs placebo (generic inverse variance method), Outcome 5 Pain (10‐cm visual analogue scale).

Comparison 1 CCBs vs placebo (generic inverse variance method), Outcome 6 Patient global.
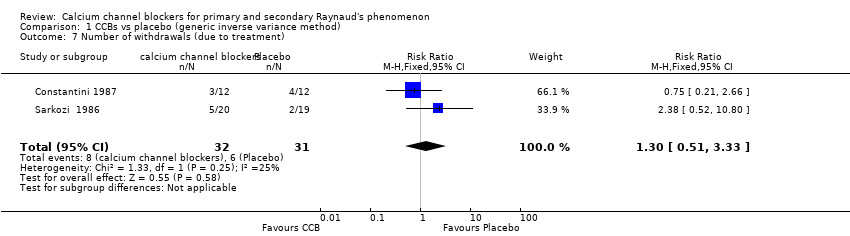
Comparison 1 CCBs vs placebo (generic inverse variance method), Outcome 7 Number of withdrawals (due to treatment).

Comparison 2 Subgroup analysis by RP type, Outcome 1 Frequency of attacks (average/week).

Comparison 2 Subgroup analysis by RP type, Outcome 2 Severity of attacks (average, on a 10‐cm VAS).

Comparison 3 Subgroup analysis: nifedipine versus placebo by RP type, Outcome 1 Frequency of attacks: nifedipine vs placebo by RP type.

Comparison 3 Subgroup analysis: nifedipine versus placebo by RP type, Outcome 2 Severity of attacks: nifedipine vs placebo by RP type.

Comparison 4 Subgroup analysis by CCB class, Outcome 1 Frequency of attacks (average/week).
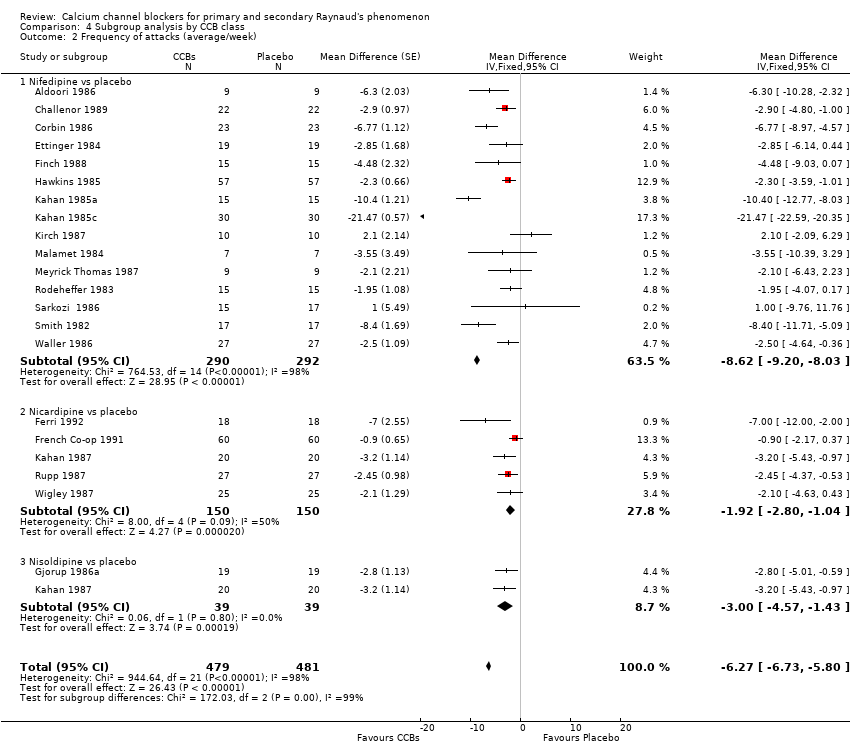
Comparison 4 Subgroup analysis by CCB class, Outcome 2 Frequency of attacks (average/week).
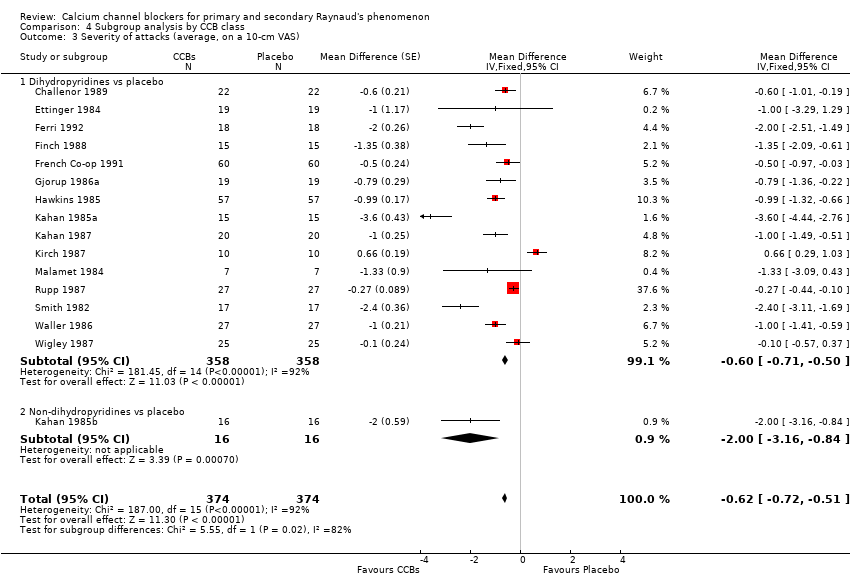
Comparison 4 Subgroup analysis by CCB class, Outcome 3 Severity of attacks (average, on a 10‐cm VAS).

Comparison 4 Subgroup analysis by CCB class, Outcome 4 Severity of attacks (average, on a 10‐cm VAS).

Comparison 5 Subgroup analysis by CCB dose, Outcome 1 Frequency of attacks (average/week).

Comparison 5 Subgroup analysis by CCB dose, Outcome 2 Duration of attacks (minutes).
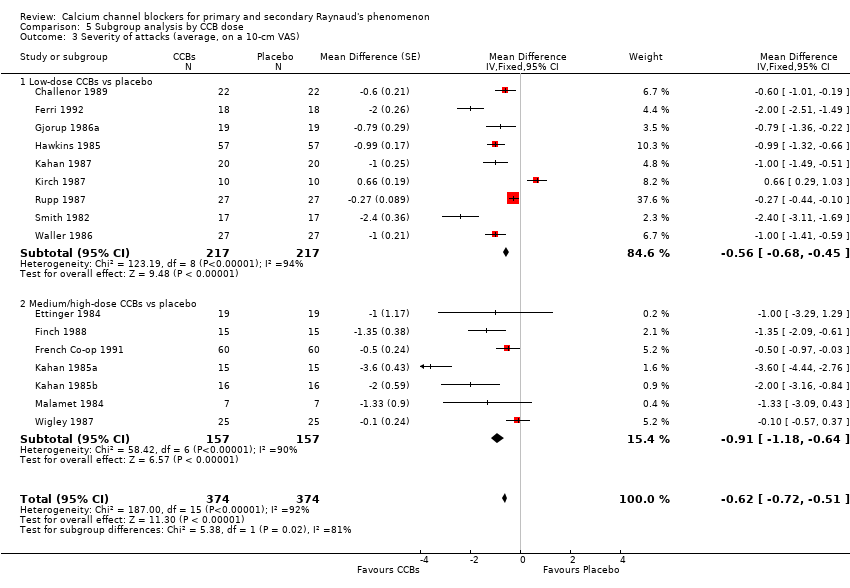
Comparison 5 Subgroup analysis by CCB dose, Outcome 3 Severity of attacks (average, on a 10‐cm VAS).
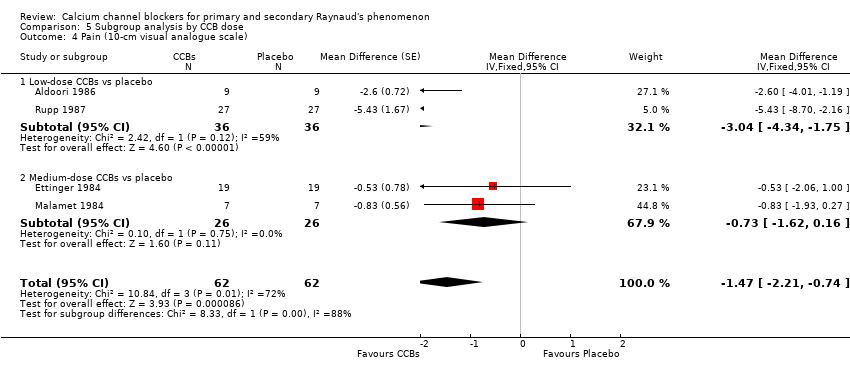
Comparison 5 Subgroup analysis by CCB dose, Outcome 4 Pain (10‐cm visual analogue scale).

Comparison 5 Subgroup analysis by CCB dose, Outcome 5 Patient global.

Comparison 6 Minor outcomes, Outcome 1 Number of participants with improvement.

Comparison 6 Minor outcomes, Outcome 2 Side effects.
| Calcium channel blockers (CCBs) compared with placebo for treatment of Raynaud's phenomenon | ||||||
| Patient or population: patients with Raynaud's phenomenon | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | CCBs (all) | |||||
| Frequency of attacks | Mean frequency of attacks in control groups: 13.7 attacksa | Mean frequency of attacks in intervention groups: 6.13 lower (6.60 to 5.67 lower)b | 528 | ⊕⊕⊕⊝ moderatec | Note: Excluding a study with a very large reduction in frequency of attacks changed the mean difference to ‐2.93 per week (95% CI ‐3.44 to ‐2.43). Absolute risk difference: N/Ad Relative percent change: ‐44% (95% CI ‐48% to ‐41%) | |
| Duration of attacks Average duration per attack measured in minutes | Mean duration of attacks in control groups: | Mean duration of attacks in intervention groups: (‐3.29 to 0) | 69 | ⊕⊕⊝⊝ lowc,e | NNTB: N/Ad Absolute risk difference: N/Ad Relative percent change: ‐9% (95% CI ‐18% to 0%) | |
| Severity of attacks | Mean severity of attacks in control groups: | Mean severity of attacks in intervention groups: | 415 | ⊕⊕⊕⊝ moderatec | NNTB: N/Ad Absolute risk difference: ‐6% (95% CI ‐7% to ‐5%) Relative percent change: ‐9% (95% CI ‐11% to ‐8%) | |
| Pain Average pain per attack, measured on a 10‐cm visual analogue scale (0 = no pain, 10 = maximal pain) | Mean pain in control groups: | Mean pain in intervention groups: | 62 | ⊕⊕⊝⊝ lowc,e | N/Ad Absolute risk difference: ‐15% (95% CI ‐22% to ‐7%) Relative percent change: ‐47% (95% CI ‐71% to ‐24%) | |
| Patient global Follow‐up: 5 weeks | Mean patient global in control group: 3.9 cma | Mean patient global in intervention groups: | 92 | ⊕⊕⊕⊝ moderatee | NNTB: N/Af Absolute risk difference: ‐4% (95% CI ‐7% to 0%) Relative percent change: ‐9% (95% CI ‐19% to 0%) | |
| Number of withdrawals due to adverse events | 194 per 1000 | 252 per 1000 | RR 1.30 | 63 | ⊕⊕⊝⊝ lowe,g | NNTH: N/Af Absolute risk reduction: 6% (95% CI ‐14% to 26%) Relative percent change: 30% (95% CI ‐49% to 233%) |
| Serious adverse events | See comment. | See comment. | Not estimable | 0 | See comment. | No serious adverse events reported |
| *The basis for the assumed risk (eg, median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aFinal value: weighted mean of scores in placebo group across studies in the meta‐analysis. | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Frequency of attacks (average/week) Show forest plot | 23 | 1024 | Mean Difference (Fixed, 95% CI) | ‐6.07 [‐6.53, ‐5.61] |
| 2 Frequency of attacks (average/week) Show forest plot | 22 | Mean Difference (Fixed, 95% CI) | ‐2.93 [‐3.44, ‐2.43] | |
| 3 Duration of attacks (minutes) Show forest plot | 6 | 138 | Mean Difference (Fixed, 95% CI) | ‐1.67 [‐3.29, ‐0.04] |
| 4 Severity of attacks (average, on a 10‐cm VAS) Show forest plot | 16 | 748 | Risk Difference (Fixed, 95% CI) | ‐0.62 [‐0.72, ‐0.51] |
| 5 Pain (10‐cm visual analogue scale) Show forest plot | 4 | 124 | Std. Mean Difference (Fixed, 95% CI) | ‐1.47 [‐2.21, ‐0.74] |
| 6 Patient global Show forest plot | 2 | 192 | Std. Mean Difference (Fixed, 95% CI) | ‐0.37 [‐0.73, ‐0.02] |
| 7 Number of withdrawals (due to treatment) Show forest plot | 2 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.51, 3.33] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Frequency of attacks (average/week) Show forest plot | 15 | 624 | Mean Difference (Fixed, 95% CI) | ‐3.15 [‐3.67, ‐2.63] |
| 1.1 CCBs vs placebo for primary RP | 12 | 420 | Mean Difference (Fixed, 95% CI) | ‐3.02 [‐3.65, ‐2.38] |
| 1.2 CCBs vs placebo for secondary RP | 9 | 204 | Mean Difference (Fixed, 95% CI) | ‐3.42 [‐4.33, ‐2.51] |
| 2 Severity of attacks (average, on a 10‐cm VAS) Show forest plot | 10 | 506 | Mean Difference (Fixed, 95% CI) | ‐0.67 [‐0.77, ‐0.57] |
| 2.1 Primary RP: CCBs vs placebo | 8 | 368 | Mean Difference (Fixed, 95% CI) | ‐0.95 [‐1.11, ‐0.79] |
| 2.2 Secondary RP: CCBs vs placebo | 6 | 138 | Mean Difference (Fixed, 95% CI) | ‐0.48 [‐0.61, ‐0.35] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Frequency of attacks: nifedipine vs placebo by RP type Show forest plot | 9 | 268 | Mean Difference (Fixed, 95% CI) | ‐4.34 [‐5.09, ‐3.59] |
| 1.1 Nifedipine vs placebo for primary RP | 6 | 148 | Mean Difference (Fixed, 95% CI) | ‐4.42 [‐5.35, ‐3.50] |
| 1.2 Nifedipine vs placebo for secondary RP | 6 | 120 | Mean Difference (Fixed, 95% CI) | ‐4.19 [‐5.47, ‐2.91] |
| 2 Severity of attacks: nifedipine vs placebo by RP type Show forest plot | 4 | 108 | Mean Difference (Fixed, 95% CI) | ‐0.82 [‐1.07, ‐0.58] |
| 2.1 Primary RP | 2 | 54 | Mean Difference (Fixed, 95% CI) | ‐1.74 [‐2.09, ‐1.39] |
| 2.2 Secondary RP | 3 | 54 | Mean Difference (Fixed, 95% CI) | 0.01 [‐0.32, 0.34] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Frequency of attacks (average/week) Show forest plot | 23 | Mean Difference (Fixed, 95% CI) | ‐6.07 [‐6.53, ‐5.61] | |
| 1.1 Dihydropyridines vs placebo | 22 | Mean Difference (Fixed, 95% CI) | ‐6.13 [‐6.60, ‐5.67] | |
| 1.2 Non‐dihydropyridines vs placebo | 1 | Mean Difference (Fixed, 95% CI) | ‐3.15 [‐6.33, 0.03] | |
| 2 Frequency of attacks (average/week) Show forest plot | 21 | 960 | Mean Difference (Fixed, 95% CI) | ‐6.27 [‐6.73, ‐5.80] |
| 2.1 Nifedipine vs placebo | 15 | 582 | Mean Difference (Fixed, 95% CI) | ‐8.62 [‐9.20, ‐8.03] |
| 2.2 Nicardipine vs placebo | 5 | 300 | Mean Difference (Fixed, 95% CI) | ‐1.92 [‐2.80, ‐1.04] |
| 2.3 Nisoldipine vs placebo | 2 | 78 | Mean Difference (Fixed, 95% CI) | ‐1.00 [‐4.57, ‐1.43] |
| 3 Severity of attacks (average, on a 10‐cm VAS) Show forest plot | 16 | 748 | Mean Difference (Fixed, 95% CI) | ‐0.62 [‐0.72, ‐0.51] |
| 3.1 Dihydropyridines vs placebo | 15 | 716 | Mean Difference (Fixed, 95% CI) | ‐0.60 [‐0.71, ‐0.50] |
| 3.2 Non‐dihydropyridines vs placebo | 1 | 32 | Mean Difference (Fixed, 95% CI) | ‐2.0 [‐3.16, ‐0.84] |
| 4 Severity of attacks (average, on a 10‐cm VAS) Show forest plot | 15 | 716 | Mean Difference (Fixed, 95% CI) | ‐0.60 [‐0.71, ‐0.50] |
| 4.1 Nifedipine vs placebo | 9 | 378 | Mean Difference (Fixed, 95% CI) | ‐0.79 [‐0.96, ‐0.61] |
| 4.2 Nicardipine vs placebo | 5 | 300 | Mean Difference (Fixed, 95% CI) | ‐0.47 [‐0.61, ‐0.33] |
| 4.3 Nisoldipine vs placebo | 1 | 38 | Mean Difference (Fixed, 95% CI) | ‐0.79 [‐1.36, ‐0.22] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Frequency of attacks (average/week) Show forest plot | 23 | 1024 | Mean Difference (Fixed, 95% CI) | ‐6.07 [‐6.53, ‐5.61] |
| 1.1 Low‐dose CCBs vs placebo | 14 | 620 | Mean Difference (Fixed, 95% CI) | ‐1.00 [‐3.63, ‐2.37] |
| 1.2 Medium/high‐dose CCBs vs placebo | 9 | 404 | Mean Difference (Fixed, 95% CI) | ‐9.50 [‐10.17, ‐8.83] |
| 2 Duration of attacks (minutes) Show forest plot | 6 | 138 | Mean Difference (Fixed, 95% CI) | ‐1.67 [‐3.29, ‐0.04] |
| 2.1 Low‐dose CCBs vs placebo | 3 | 56 | Mean Difference (Fixed, 95% CI) | 2.24 [‐0.24, 4.73] |
| 2.2 Medium‐dose CCBs vs placebo | 3 | 82 | Mean Difference (Fixed, 95% CI) | ‐4.60 [‐6.76, ‐2.45] |
| 3 Severity of attacks (average, on a 10‐cm VAS) Show forest plot | 16 | 748 | Mean Difference (Fixed, 95% CI) | ‐0.62 [‐0.72, ‐0.51] |
| 3.1 Low‐dose CCBs vs placebo | 9 | 434 | Mean Difference (Fixed, 95% CI) | ‐0.56 [‐0.68, ‐0.45] |
| 3.2 Medium/high‐dose CCBs vs placebo | 7 | 314 | Mean Difference (Fixed, 95% CI) | ‐0.91 [‐1.18, ‐0.64] |
| 4 Pain (10‐cm visual analogue scale) Show forest plot | 4 | 124 | Mean Difference (Fixed, 95% CI) | ‐1.47 [‐2.21, ‐0.74] |
| 4.1 Low‐dose CCBs vs placebo | 2 | 72 | Mean Difference (Fixed, 95% CI) | ‐3.04 [‐4.34, ‐1.75] |
| 4.2 Medium‐dose CCBs vs placebo | 2 | 52 | Mean Difference (Fixed, 95% CI) | ‐0.73 [‐1.62, 0.16] |
| 5 Patient global Show forest plot | 2 | 192 | Mean Difference (Fixed, 95% CI) | ‐0.37 [‐0.73, ‐0.02] |
| 5.1 Low‐dose CCBs vs placebo | 1 | 72 | Mean Difference (Fixed, 95% CI) | ‐0.2 [‐0.63, 0.23] |
| 5.2 High‐dose CCBs vs placebo | 1 | 120 | Mean Difference (Fixed, 95% CI) | ‐0.74 [‐1.37, ‐0.11] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Number of participants with improvement Show forest plot | 3 | 59 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.38 [1.35, 4.20] |
| 2 Side effects Show forest plot | 3 | 78 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.87, 1.45] |