Benzodiazepines for antipsychotic‐induced tardive dyskinesia
Abstract
Background
Tardive dyskinesia (TD) is a disfiguring movement disorder, often of the orofacial region, frequently caused by using antipsychotic drugs. A wide range of strategies have been used to help manage TD, and for those who are unable to have their antipsychotic medication stopped or substantially changed, the benzodiazepine group of drugs have been suggested as a useful adjunctive treatment. However, benzodiazepines are very addictive.
Objectives
To determine the effects of benzodiazepines for antipsychotic‐induced tardive dyskinesia in people with schizophrenia, schizoaffective disorder, or other chronic mental illnesses.
Search methods
On 17 July 2015 and 26 April 2017, we searched the Cochrane Schizophrenia Group's Study‐Based Register of Trials (including trial registers), inspected references of all identified studies for further trials and contacted authors of each included trial for additional information.
Selection criteria
We included all randomised controlled trials (RCTs) focusing on people with schizophrenia (or other chronic mental illnesses) and antipsychotic‐induced TD that compared benzodiazepines with placebo, no intervention, or any other intervention for the treatment of TD.
Data collection and analysis
We independently extracted data from the included studies and ensured that they were reliably selected, and quality assessed. For homogenous dichotomous data, we calculated random effects, risk ratio (RR), and 95% confidence intervals (CI). We synthesised continuous data from valid scales using mean differences (MD). For continuous outcomes, we preferred endpoint data to change data. We assumed that people who left early had no improvement.
Main results
The review now includes four trials (total 75 people, one additional trial since 2006, 21 people) randomising inpatients and outpatients in China and the USA. Risk of bias was mostly unclear as reporting was poor. We are uncertain about all the effects as all evidence was graded at very low quality. We found no significant difference between benzodiazepines and placebo for the outcome of 'no clinically important improvement in TD' (2 RCTs, 32 people, RR 1.12, 95% CI 0.60 to 2.09, very low quality evidence). Significantly fewer participants allocated to clonazepam compared with phenobarbital (as active placebo) experienced no clinically important improvement (RR 0.44, 95% CI 0.20 to 0.96, 1 RCT, 21 people, very low quality evidence). For the outcome 'deterioration of TD symptoms,' we found no clear difference between benzodiazepines and placebo (2 RCTs, 30 people, RR 1.48, 95% CI 0.22 to 9.82, very low quality evidence). All 10 participants allocated to benzodiazepines experienced any adverse event compared with 7/11 allocated to phenobarbital (RR 1.53, 95% CI 0.97 to 2.41, 1 RCT, 21 people, very low quality evidence). There was no clear difference in the incidence of participants leaving the study early for benzodiazepines compared with placebo (3 RCTs, 56 people, RR 2.73, 95% CI 0.15 to 48.04, very low quality evidence) or compared with phenobarbital (as active placebo) (no events, 1 RCT, 21 people, very low quality evidence). No trials reported on social confidence, social inclusion, social networks, or personalised quality of life, which are outcomes designated important by patients. No trials comparing benzodiazepines with placebo or treatment as usual reported on adverse effects.
Authors' conclusions
There is only evidence of very low quality from a few small and poorly reported trials on the effect of benzodiazepines as an adjunctive treatment for antipsychotic‐induced TD. These inconclusive results mean routine clinical use is not indicated and these treatments remain experimental. New and better trials are indicated in this under‐researched area; however, as benzodiazepines are addictive, we feel that other techniques or medications should be adequately evaluated before benzodiazepines are chosen.
PICO
Plain language summary
Benzodiazepines for antipsychotic‐induced tardive dyskinesia
Review question
To determine the effectiveness of benzodiazepines in the treatment of tardive dyskinesia in people with schizophrenia or other similar mental health problems.
Background
People with schizophrenia often hear voices and see things (hallucinations), and have strange beliefs (delusions). The main treatment for schizophrenia is antipsychotic drugs. However, these drugs can have debilitating side effects. Tardive dyskinesia is an involuntary (uncontrollable and unintended) movement that causes the face, mouth, tongue, and jaw to convulse, spasm, and grimace. It is caused by prolonged or high‐dose use of antipsychotic drugs, is difficult to treat, and can be incurable. The benzodiazepine group of medicines have been suggested as a useful add‐on treatment for tardive dyskinesia. However, benzodiazepines are very addictive.
Study characteristics
The review includes four clinical trials with 75 people who had tardive dyskinesia as a result of using antipsychotic medicines. The participants were randomised into groups that received either their usual antipsychotic medicine plus a benzodiazepine or their usual antipsychotic plus a placebo (dummy medicine).
Key results
Improvement in TD symptoms was similar between the treatment groups. Participants were just as likely to leave the studies early from the placebo groups as the benzodiazepine groups. Data were not available for outcomes important to patients such as improvement in social confidence, social inclusion, social networks or quality of life.
Quality of the evidence
Evidence is limited because the trials are so few, small, and poorly reported. It is uncertain whether benzodiazepines are helpful in the treatment of tardive dyskinesia. The use of benzodiazepines for treating people with antipsychotic‐induced TD therefore remains experimental, and because they are highly addictive, a last resort. The low number of studies in this review strongly indicates that this is not an active area of research. To fully investigate whether benzodiazepines have any positive effects for people with tardive dyskinesia, there would have to be more well‐designed, conducted and reported trials.
This plain language summary was adapted by the review authors from a summary originally written by Ben Gray, Senior Peer Researcher, McPin Foundation (mcpin.org/).
Authors' conclusions
Summary of findings
| Benzodiazepines compared with placebo for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: psychiatric patients (mainly schizophrenia) with antipsychotic‐induced tardive dyskinesia | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Risk with placebo/no treatment | Risk with benzodiazepines | |||||
| Tardive dyskinesia: no clinically important improvement | Study population | RR 1.12 | 32 | ⊕⊝⊝⊝ | ‐ | |
| 545 per 1000 | 611 per 1000 | |||||
| Tardive dyskinesia: deterioration in symptoms | Study population | RR 1.48 | 30 | ⊕⊝⊝⊝ | ‐ | |
| 91 per 1000 | 135 per 1000 | |||||
| Adverse effect: any adverse event | None of the included studies reported on these outcomes. | |||||
| Adverse effect: no clinically significant extrapyramidal adverse effects | ||||||
| Acceptability of the treatment (measured by participants leaving the study early) | Study population | RR 2.73 | 56 | ⊕⊝⊝⊝ | ‐ | |
| 0 per 1000 | 0 per 1000 | |||||
| Social confidence, social inclusion, social networks, or personalised quality of life ‐ not reported | None of the included studies reported on this outcome. | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded one level for risk of bias: none of the studies adequately described randomisation procedure or allocation concealment, one study did not blind participants and personnel, and one study was a post hoc subgroup analysis of participants with tardive dyskinesia. 2Downgraded two levels for imprecision: small sample size, and 95% CI of effect estimate includes both appreciable benefit and appreciable harm for benzodiazepines. | ||||||
| Benzodiazepines compared with phenobarbital (as active placebo) for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: psychiatric patients (mainly schizophrenia) with antipsychotic‐induced tardive dyskinesia | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Risk with phenobarbital (active placebo) | Risk with benzodiazepines | |||||
| Tardive dyskinesia: no clinically important improvement | Study population | RR 0.44 | 21 | ⊕⊝⊝⊝ | ‐ | |
| 909 per 1000 | 400 per 1000 | |||||
| Tardive dyskinesia: deterioration in symptoms ‐ not measured | The included study did not report on this outcome. | |||||
| Adverse events: any | Study population | RR 1.53 | 21 | ⊕⊝⊝⊝ | ‐ | |
| 636 per 1000 | 974 per 1000 | |||||
| Adverse effect: extrapyramidal symptoms ‐ not reported | The included study did not report on this outcome. | |||||
| Acceptability of the treatment (measured by participants leaving the study early) | Study population | Not estimable | 21 | ⊕⊝⊝⊝ | No events were reported; no one left the study early. | |
| 0 per 1000 | 0 per 1000 | |||||
| Social confidence, social inclusion, social networks, or personalised quality of life ‐ not measured | The included study did not report on this outcome. | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded one level for risk of bias: the included study did not adequately describe randomisation procedure, allocation concealment, or blinding. 2Downgraded two levels for imprecision: only one study with a very small sample size. | ||||||
Background
Description of the condition
The management of schizophrenia and other chronic mental illnesses was revolutionised in the 1950s with the introduction of antipsychotic (or neuroleptic) medications. These medications are effective in the control of symptoms such as abnormal perceptions (hallucinations), disordered thoughts (impaired communication), and fixed false beliefs (delusions) (Donlon 1980). When used as a maintenance therapy for schizophrenia, antipsychotic drugs are associated with a reduced risk of relapse (Schooler 1993). However, antipsychotic medications have been associated with a range of adverse effects that can affect quality of life and lead to poor compliance with treatment, such as tardive dyskinesia (TD) (Barnes 1993).
TD is a chronic condition of insidious onset, characterised by abnormal, repetitive, and involuntary movements (APA 1992). The clinical features include tongue protrusion, side‐to‐side or rotatory movement of the jaw, lip smacking, puckering and pursing, and rapid eye blinking (Casey 1994). In some people, rapid movements of the arms, legs, and trunk may also occur. In long‐term studies, antipsychotic medications have been associated with an incidence of TD of approximately 5% per year in adults and 25% to 30% in elderly people (Correll 2004). Studies on the natural history of TD have reported widely variable remission rates (1% to 62%) depending on a person's age, psychiatric diagnosis, course of the psychiatric disorder, and duration of therapy (Bergen 1989; Fernandez 2001; Glazer 1990).
The prevalence of TD is often thought to be decreasing based on the use of second‐generation antipsychotic drugs (SGA) in place of first‐generation antipsychotic drugs (FGA) (Cloud 2014). One systematic review found that the incidence of TD associated with SGA (2% to 4%) was significantly lower than that for FGA (5% to 8%) (Correll 2008). In older adults, the risk is reported to be more than three times lower when receiving SGA after one year of treatment (O'Brien 2016). Despite this, this widespread use of SGA in clinical settings may still result in an overall increase in the number of cases of TD (Glazer 2000a).
Although the most frequent cause of TD is the use of antipsychotic medication, it is striking that dose reduction can lead to a temporary exacerbation in symptoms. Conversely, increasing the dose is often associated with a temporary remission. Antipsychotic drugs block certain chemical receptor sites in the brain ‐ one of these is specific for dopamine (Casey 1994). One hypothesis explaining the cause of antipsychotic‐induced TD is that chronic blockade of dopamine receptors in specific cells of the brain (neurons from the nigrostriatum) causes an overgrowth of these receptors (Casey 1994). However, there is some suggestion that the chronic use of antipsychotic drugs may also cause an abnormal production of highly active atoms and chemical groups (cytotoxic free radicals), which may damage specific cells in the brain. This, in turn, could be responsible for the appearance of TD (Cadet 1989).
Description of the intervention
After the discovery of chlordiazepoxide in the late 1950s by Leo Sternbach, benzodiazepines became widely available and were prescribed to hundreds of millions of people in various medical settings (Dell'Osso 2015). Their high therapeutic index, the availability of the antagonist flumazenil in case of overdose, and their rapid onset of action make these compounds particularly versatile and difficult to replace in clinical psychiatry. Benzodiazepines are the pharmacological mainstay of the clinical management of anxiety and sleep disorders, but are commonly used as an adjunctive treatment for psychotic disorders and schizophrenia, particularly when people display agitated, violent, and aggressive behaviours (Dell'Osso 2015).
How the intervention might work
Benzodiazepines have been included as a candidate treatment for TD in several practice guidelines (APA 1992; Gardos 1994; Jeste 1988). It has been suggested that the chronic blockade of dopamine receptors in TD leads to inactivity in another set of cells that employ gamma‐aminobutyric‐acid (GABA) (Barnes 1993). The benzodiazepine group of drugs are the most widely used GABA agonists and will be the focus of this review. There is limited evidence from animal experiments to suggest that GABA dysfunction is also associated with movement disorders (Gunne 1984).
Why it is important to do this review
Several SGA have been produced in recent decades that claim to cause less or no TD (Lieberman 1996). These claims may or may not be true, and certainly evidence does suggest that thoughtful use of older‐generation drugs is not associated with more TD than with newer treatments (Chouinard 2008). However, in a global context, it is likely that the less expensive and more familiar drugs (such as chlorpromazine or haloperidol) will continue to be the mainstay of treatment of people with schizophrenia (WHO Essential List 2010). Use of drugs such as these is associated with emergence of TD and, therefore, this condition will remain a problem for years to come.
TD can result in considerable social and physical disability (Barnes 1993), and symptoms are often irreversible (Bergen 1989; Fernandez 2001; Gerlach 1988; Glazer 1990). Additionally, TD is frequently associated with lower quality of life (Ascher‐Svanum 2008) and a greater mortality rate (Chong 2009). Given the high incidence and prevalence of TD among people taking antipsychotic medication, the need for prevention or treatment is clear. Unfortunately, there has been sparse evidence to guide clinicians (NICE 2014; Taylor 2009). Although many treatments have been tested, no one intervention has been shown clearly to be effective. Cessation or reduction of the dose of antipsychotic medication is the ideal management for TD. In clinical practice this is not always possible, not least because in many people such a reduction would lead to relapse. This review focused on whether the addition of benzodiazepine treatments to people already receiving antipsychotic medication is likely to help TD.
This review is one in a series of Cochrane Reviews (see Table 1) evaluating treatments for antipsychotic‐induced TD, and is an update of a Cochrane Review first published in 1999 (Soares‐Weiser 1999), and previously updated in 2003 (Walker 2003) and in 2006 (Bhoopathi 2006).
| Interventions | Reference |
| Anticholinergic medication | Soares‐Weiser 1997; Soares‐Weiser 2000; 2016 update to be published. |
| Benzodiazepines | This review. |
| Calcium channel blockers | Essali 2011; 2016 update to be published. |
| Cholinergic medication | Tammenmaa 2002; 2016 update to be published. |
| Gamma‐aminobutyric acid agonists | Alabed 2011; 2016 update to be published. |
| Miscellaneous treatments | Soares‐Weiser 2003; 2016 update to be published. |
| Neuroleptic reduction or cessation (or both) and neuroleptics | Soares‐Weiser 2006; 2016 update to be published. |
| Non‐neuroleptic catecholaminergic drugs | El‐Sayeh 2006; 2016 update to be published. |
| Vitamin E | Soares‐Weiser 2011; 2016 update to be published. |
Objectives
To determine the effects of benzodiazepines for antipsychotic‐induced tardive dyskinesia in people with schizophrenia, schizoaffective disorder, or other chronic mental illnesses.
Methods
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials (RCTs). We included trials that implied randomisation if they were described as 'double‐blind' and the demographic details of each group were similar. We excluded quasi‐randomised studies, such as those allocated by using alternate days of the week.
Types of participants
We included people with schizophrenia, schizoaffective disorder, or other serious chronic mental illness diagnosed by any criteria, irrespective of gender, age or nationality that:
-
required the use of antipsychotics for more than three months; and
-
developed TD (diagnosed by any criteria) during antipsychotic treatment; and
-
for whom the dose of antipsychotic medication had been stable for one month or more (the same applied for participants free of antipsychotic drugs).
Types of interventions
1. The benzodiazepine family of drugs
Alprazolam, bromazepam, chlordiazepoxide, clobazam, clonazepam, clorazepate dipotassium, diazepam, flunitrazepam, flurazepam, loprazolam, lorazepam, lormetazepam, medazepam, midazolam, nitrazepam, oxazepam, temazepam at any dose or means of administration, compared with:
a. Placebo or no intervention; or
b. Any other intervention for the treatment of tardive dyskinesia
Types of outcome measures
We defined clinical efficacy as an improvement in the symptoms of TD of more than 50%, on any scale. We grouped outcomes into short term (less than six weeks), medium term (between six weeks and six months) and long term (more than six months).
Primary outcomes
1. Tardive dyskinesia
No clinically important improvement in symptoms, defined as more than 50% improvement on any TD scale at any time period.a
2. Adverse effects
No clinically significant extrapyramidal adverse effects at any time period.
aThe primary outcome for previous versions of this review was 'any improvement in TD symptoms of more than 50% on any TD scale ‐ any time period.' Data provided in trials did not fit this exactly; however, we felt 'not improved to a clinically important extent' fit best with what we had hoped to find.
Secondary outcomes
1. Tardive dyskinesia
1.1 Any improvement in symptoms on any TD scale, as opposed to no improvement.
1.2 Deterioration in symptoms, defined as any deleterious change on any TD scale.
1.3 Mean change in severity of TD during the trial period.
1.4 Mean difference in severity of TD at the end of the trial.
2. General mental state changes
2.1 Deterioration in general psychiatric symptoms (such as delusions and hallucinations) defined as any deleterious change on any scale.
2.2 Mean difference in severity of psychiatric symptoms at the end of the trial.
3. Acceptability of the treatment
3.1 Acceptability of the intervention to the participant group as measured by numbers of people leaving the trial early.
4. Adverse effects
4.1 Use of any anti‐parkinsonism drugs.
4.2 Mean score/change in extrapyramidal adverse effects.
4.3 Acute dystonia.
5. Other adverse effects, general and specific
6. Hospital and service utilisation outcomes
6.1 Hospital admission.
6.2 Mean change in days in hospital.
6.3 Improvement in hospital status (e.g. change from formal to informal admission status, use of seclusion, level of observation).
7. Economic outcomes
7.1 Mean change in total cost of medical and mental health care.
7.2 Total indirect and direct costs.
8. Social confidence, social inclusion, social networks, or personalised quality of life measures
8.1 No significant change in social confidence, social inclusion, social networks, or personalised quality of life measures.
8.2 Mean score/change in social confidence, social inclusion, social networks, or personalised quality of life measures.
9. Behaviour
9.1 Clinically significant agitation.
9.2 Use of adjunctive medication for sedation.
9.3 Aggression to self or others.
10. Cognitive state
10.1 No clinically important change.
10.2 No change, general and specific.
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011) and used GRADEpro to export data from this review to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of the available data on all outcomes we rated as important to patient care and decision making. This summary was used to guide our conclusions. We selected the following main outcomes for inclusion in the 'Summary of findings' tables:
1. Tardive dyskinesia
1.1 No clinically important improvement in symptoms, defined as more than 50% improvement on any TD scale.
1.2 Deterioration.
2. Adverse effect
2.1 Any adverse event.
2.2 No clinically significant extrapyramidal adverse effects.
3. Acceptability of treatment
3.1 Leaving the study early.
4. Social confidence, social inclusion, social networks, or personalised quality of life measuresb
4.1 No significant change in social confidence, social inclusion, social networks, or personalised quality of life measures for either recipients of care or carers.
bOutcome designated important to patients. We wished to add perspectives from people's personal experience with TD to the research agenda. A consultation with service users was planned where a previously published version of a review in the Cochrane TD series (Soares‐Weiser 2011; Table 1) and a lay overview of that review gave the foundation for the discussions. The session was planned to provide time to reflect on current research on TD and to consider gaps in knowledge. The report is not completed but we will add link to it within this review but have added one figure showing service user expression of frustration concerning this neglected area of research (Figure 1). Informed by the results of the consultation, for this review, we updated the list of outcomes and included outcomes for the 'Summary of findings' table.

Message from one of the participants of the public and patient involvement consultation of service user perspectives on tardive dyskinesia research.
Search methods for identification of studies
Electronic searches
The 2015 and 2017 update searches were carried out in parallel with updating eight other TD reviews, see Table 1 for details. The search covered all nine TD reviews. For previous searches, see Appendix 1.
Cochrane Schizophrenia Group's Study‐Based Register of Trials
On 16 July 2015 and 26 April 2017, the information specialist searched the register using the following search strategy:
*Tardive Dyskinesia* in Health Care Condition Field of STUDY
In such a study‐based register, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics (Shokraneh 2017).
This register is compiled by systematic searches of major resources (AMED, BIOSIS, CINAHL, ClinicalTrials.Gov, Embase, MEDLINE, PsycINFO, PubMed, World Health Organization (WHO) ICTRP) and their monthly updates, ProQuest Dissertations and Theses A&I and its quarterly update, Chinese databases (CBM, CNKI, and Wanfang) and their annual updates, handsearches, grey literature, and conference proceedings (see Group's Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
Searching other resources
1. Reference searching
We inspected references of all identified studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Methods used for the 2017 update are presented below, the methods used in the previous versions are in Appendix 1.
Selection of studies
Two review authors (RA and AG) inspected all abstracts of studies identified and potentially relevant reports. We resolved disagreement by discussion, or, where there was still doubt, we acquired the full article for further inspection. We acquired the full articles of relevant reports/abstracts meeting initial criteria for reassessment and carefully inspected for a final decision on inclusion (see Criteria for considering studies for this review). The two review authors (RA and AG) were not blinded to the names of the authors, institutions or journal of publication. Where difficulties or disputes arose, we asked a third review author (HB) for help and where it was impossible to decide or if adequate information was not available to make a decision, we added these studies to those awaiting assessment and contacted the authors of the papers for clarification.
Data extraction and management
1. Extraction
Two review authors (RA and HB) independently extracted data from all included studies. We discussed any disagreements and documented decisions. With remaining problems, one review author (KSW) helped clarify issues and we documented these final decisions. We extracted data presented only in graphs and figures whenever possible, but included them only if two review authors independently had the same result. We attempted to contact authors through an open‐ended request to obtain missing information or for clarification whenever necessary. If studies were multicentre, where possible, we extracted data relevant to each component centre separately.
2. Management
2.1 Forms
We extracted data online in Covidence.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
-
the psychometric properties of the measuring instrument were described in a peer‐reviewed journal (Marshall 2000); and
-
the measuring instrument had not been written or modified by one of the trialists for that particular trial.
Ideally the measuring instrument should have been either a self‐report or completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, we noted in Description of studies if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. In contrast, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult‐to‐measure conditions such as schizophrenia. We decided to primarily use endpoint data, and only use change data if endpoint data were not available. We combined endpoint and change data in the analysis as we preferred to use mean differences (MD) rather than standardised mean differences throughout (Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to relevant data before inclusion.
Please note, we entered data from studies of at least 200 participants in the analysis, because skewed data pose less of a problem in large studies. We also entered all relevant change data as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not.
For endpoint data from studies with fewer than 200 participants.
-
When a scale started from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation (SD). If this value was lower than 1, it strongly suggested a skew and we excluded these data. If this ratio was higher than 1 but below 2, there was suggestion of skew. We entered these data and tested whether their inclusion or exclusion changed the results substantially. Finally, if the ratio was larger than 2, we included these data, because skew was less likely (Altman 1996; Higgins 2011).
-
If a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS) (Kay 1986)), which can have values from 30 to 210), we modified the calculation described in (1) above to take the scale starting point into account. In these cases, skew was present if 2 SD > (S ‐ Smin), where S was the mean score and Smin was the minimum score.
2.5 Common measure
Where relevant, to facilitate comparison between trials, we converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week, or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we converted continuous outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved.' It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986), this can be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for benzodiazepines. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved'), we presented data where the left of the line indicated an unfavourable outcome and noted this in the relevant graphs.
Assessment of risk of bias in included studies
Two review authors (RA and HB) independently assessed risk of bias within the included studies by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions to assess trial quality (Higgins 2011). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data, and selective reporting.
If the raters disagreed, we made the final rating by consensus. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies to obtain further information. If non‐concurrence occurred, we reported this.
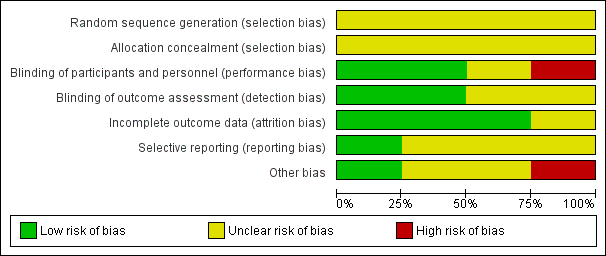
We noted the level of risk of bias in the text of the review and in Figure 2; Figure 3 and summary of findings Table for the main comparison; summary of findings Table 2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios as odds ratios tend to be interpreted as RR by clinicians (Deeks 2000).
2. Continuous data
For continuous outcomes, we estimated MD between groups. We preferred not to calculate effect size measures (standardised mean difference). However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, CIs unduly narrow, and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If any of the included trials had randomised participants by clusters, and where clustering was not accounted for in primary studies, we would have presented such data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intra‐class correlation coefficients for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will present these data as if from a non‐cluster randomised study, but adjust for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect.' This is calculated using the mean number of participants per cluster (m) and the intra‐class correlation coefficient (ICC) (design effect = 1 + (m ‐ 1) × ICC) (Donner 2002). If the ICC was not reported, we assumed it to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies would be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological, or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, we presented the additional treatment arms in comparisons. If data were binary, we simply added and combined within the two‐by‐two table. If data were continuous, we combined data following the formula in Section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We did not use data where the additional treatment arms were not relevant.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses. However, if more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we addressed this within the 'Summary of findings' tables by down‐rating quality. We also downgraded quality within the 'Summary of findings' tables where loss was 25% to 50% in total.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). We assumed all participants leaving the study early had no improvement. We undertook a sensitivity analysis testing how prone the primary outcomes were to change by comparing data only from people who completed the study to that point to the ITT analysis using the above assumptions.
3. Continuous
3.1 Attrition
We reported and used data where attrition for a continuous outcome was between 0% and 50%, and reported data only from people who completed the study to that point.
3.2 Standard deviations
If SDs were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and CIs available for group means, and either P value or t value available for MDs, we calculated them according to the rules described in the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011): when only the SE was reported, SDs were calculated by the formula SD = SE × square root (n). Sections 7.7.3 and 16.1.3 of the Cochrane Handbook for Systemic reviews of Interventions present detailed formulae for estimating SDs from P values, t or F values, CIs, ranges, or other statistics (Higgins 2011). If these formulae did not apply, we calculated the SDs according to a validated imputation method which was based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study's outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who left the trials early or were lost to follow‐up. Some trials just present the results of study completers, other trials use the method of last observation carried forward (LOCF), while more recently methods such as multiple imputation or mixed effects models for repeated measurements (MMRM) have become more of a standard. While MMRMs seem to be somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups is often the core problem in randomised schizophrenia trials. Therefore, we did not exclude studies based on the statistical approach used. However, we preferred to use the more sophisticated approaches. (e.g. MMRM or multiple‐imputation) and only presented completer analyses if some type of ITT data were not available. Moreover, we addressed this issue in the item "incomplete outcome data" of the 'Risk of bias' tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations that we had not predicted would arise and discussed in the text if they arose.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods that we had not predicted would arise and discussed in the text if they arose.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. The I2 statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of the I2 statistic depends on (1) the magnitude and direction of effects and (2) the strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a CI for the I2 statistic). An I2 estimate of 50% or greater accompanied by a statistically significant Chi2 statistic, can be interpreted as evidence of substantial levels of heterogeneity (Section 9.5.2 of the Cochrane Handbook for Systematic Reviews of InterventionsHiggins 2011). We explored and discussed in the text potential reasons for substantial levels of heterogeneity (see Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In future versions of this review, if funnel plots are possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. However, there is a disadvantage to the random‐effects model as it puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose the fixed‐effect model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Primary outcomes
We anticipated one subgroup analysis to test the hypothesis that the use of benzodiazepines is most effective for people with early‐onset TD (less than five years). We had hoped to present data for this subgroup for the primary outcomes.
1.2 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of benzodiazepines for people with schizophrenia in general. In addition, however, we tried to report data on subgroups of people in the same clinical state, stage, and with similar problems.
2. Investigation of heterogeneity
We reported when inconsistency was high. First, we investigated whether data were entered correctly. Second, if data were correct, we visually inspected the graph and successively removed studies outside of the company of the rest to see if homogeneity was restored. For this review, we decided that should this have occurred with data contributing to the summary finding of no more than about 10% of the total weighting, we would have presented data. If not, we did not pool such data and discussed issues. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious, we simply discussed the heterogeneity. We did not undertake sensitivity analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
If trials were described in some way as to imply randomisation, we undertook sensitivity analyses for the primary outcomes. We included these studies in the analyses and, if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we used relevant data from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption compared with completer data only. If there was a substantial difference, we reported and discussed these results but continued to employ our assumption.
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings on primary outcomes when we used our assumption compared with completer data only. We undertook a sensitivity analysis testing how prone results were to change when 'completer' data only were compared to the imputed data using the above assumption. If there was a substantial difference, we reported and discussed these results but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that we judged at high risk of bias across one or more of the domains of randomisation (see also Assessment of risk of bias in included studies) for the meta‐analysis of the primary outcomes. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, we included data from these trials in the analyses.
4. Imputed values
Had cluster trials been included, we would have undertaken a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect.
If we found substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed and random effects
We synthesised data using a fixed‐effect model; however, we also synthesised data for the primary outcomes only using a random‐effects model to evaluate whether this altered the significance of the results.
Results
Description of studies
Results of the search
The 2015 and 2017 searches were part of a search to update nine Cochrane Reviews on TD (Table 1).
Searches up to 2017 retrieved 704 references for 344 studies and had no date limitation(see Figure 4 for study flow diagram). We identified 18 potentially relevant studies for this review for which we conducted full‐text screenings. Agreement about which reports may have been randomised was 100%. Searches up to 2015 found three studies that could be included. From the 2015 search, one report was added as a new included study to this review (Bobruff 1981). This study was previously excluded because a benzodiazepine was compared to an active substance (phenobarbital as active placebo); however, for this update, we decided to include studies with active comparison groups (see Differences between protocol and review).

Study flow diagram.
The 2017 search found eight records (five studies). Editorial base of Cochrane Schizophrenia screened these records and no new studies were relevant to this review. They could be relevant to another review in this series of TD reviews (see Table 1), and have been put into awaiting assessment of Soares‐Weiser 2003.
Four studies (five references) are now included in this review (Bobruff 1981; Csernansky 1988; Weber 1983; Xiang 1997). Thirteen studies were excluded.
Included studies
Overall, the review now includes four studies published between 1981 and 1997 with 75 participants.
1. Methods
All studies were stated to be randomised and three studies reported being double blind (Bobruff 1981; Csernansky 1988; Xiang 1997). Weber 1983 did not blind participants and personnel. For further details, see Allocation (selection bias) and Blinding (performance bias and detection bias).
2. Design
Three studies presented a parallel longitudinal design (Bobruff 1981; Csernansky 1988; Xiang 1997), while one study used a cross‐over design with two periods (Weber 1983). For this study, only the data from before the first cross‐over was used for the reasons outlined above (see Unit of analysis issues).
3. Duration
Three of the studies were of medium duration (more than six weeks to up to six months). However, one study did not specifically report the duration of intervention (Bobruff 1981).
4. Participants
Participants, now totalling 75 people, were mostly men in their 50s, with diagnoses of various chronic psychiatric disorders, but mainly schizophrenia. Three studies reported that participants had antipsychotic‐induced TD diagnosed using the Abnormal Involuntary Movement Side Effects Scale (AIMS) (Bobruff 1981; Weber 1983; Xiang 1997). Csernansky 1988 included participants with significant dyskinesia rated on the Gerlach Dyskinesia Scale (GDS). The number of participants ranged from 13 to 24 (median 20).
5. Setting
The studies were conducted in a mixture of inpatient (Weber 1983; Xiang 1997) or outpatient (Csernansky 1988) settings. Bobruff 1981 did not report setting. Three of the included studies were based in the USA (Bobruff 1981; Csernansky 1988; Weber 1983), and one in China (Xiang 1997).
6. Interventions
All studies used benzodiazepines as an adjunct therapy to the standard treatment already being received by the participants. The underlying antipsychotic medications were not described by any of these studies. Weber 1983 compared diazepam (6‐25 mg/day, mean 12 mg/day) with no additional treatment to standard care. Csernansky 1988 compared diazepam (mean stable dose 48.3 mg/day) with alprazolam (mean stable dose 7.2 mg/day) and with placebo. Bobruff 1981 compared clonazepam (mean dose 3.9 mg/day) to phenobarbital (as an active placebo, mean dose 88.6 mg/day). Xiang 1997 compared clonazepam (4‐6 mg/day) with placebo.
7. Outcomes
7.1 General
Some continuous outcomes could not be extracted due to missing number of participants or missing means, SDs, or SEs. All included studies used the LOCF strategy for the ITT analysis of dichotomous data. All the studies reported changes in TD. Only Weber 1983 reported mental state changes and only Bobruff 1981 reported on adverse effects.
7.2 Scales used to measure the tardive dyskinesia symptoms
We present details of the scales that provided usable data below. We provided reasons for exclusions of data under 'Outcomes' in the Characteristics of included studies table. The four studies reported changes in the TD as measured with well‐recognised rating scales. Bobruff 1981; Weber 1983; and Xiang 1997 employed the AIMS scale, while Csernansky 1988 used the GDS.
7.2.1 Abnormal Involuntary Movement Side Effects Scale
The AIMS (Guy 1976) is a 12‐item scale consisting of a standardised examination followed by questions rating the orofacial, extremity, and trunk movements, as well as three global measurements. Each of these 10 items can be scored from 0 (none) to 4 (severe). Two additional items assess dental status. The AIMS ranges from 0 to 40, with higher scores indicating greater severity.
7.2.2 Gerlach Dyskinesia Scale
The GDS (Casey 1988) is a six‐item scale that scores up to 24. The scale is rated twice, once while the person is passive and once while active, on, for example, a standardised writing task.
7.3 Mental state changes
Only Weber 1983 reported on mental state, using the BPRS.
7.3.1 Brief Psychiatric Rating Scale
The BPRS (Overall 1962) is a brief rating scale used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. The scale has 16 items, and each item can be defined on a eight‐point scale varying from 0 (not present) to 7 (extremely severe). Scoring ranges from 24 to 168 with higher scores indicating greater severity.
Excluded studies
There were 13 excluded studies. Seven studies were not randomised (Ginsberg 2003; Jus 1974; Sarbulescu 1986; Singh 1980; Singh 1982; Singh 1983; Wang 2002). The remaining trials were randomised, but three studies did not include participants with TD (Petit 1994; Sachdev 1993; Wang 2000), Wonodi 2004 did not randomise benzodiazepines, and Thaker 1990 did not report any usable data and the study authors confirmed data were not retrievable. Finally, we excluded astudy from 1971 that did not report any usable data (Godwin‐Austen 1971); we were unable to identify up‐to‐date contact details of the study authors and we assume it very unlikely to receive a reply with data so many years later.
Studies awaiting classification
There are currently no studies awaiting classification.
Ongoing studies
We identified no ongoing studies.
Risk of bias in included studies
Refer to Figure 2 and Figure 3 for graphical overviews of the risk of bias in the included studies.
Allocation
All four included studies had an unclear risk of selection bias. While all studies stated that interventions were allocated at random, none were explicit about the methods used for sequence generation or allocation concealment.
Blinding
Three studies were conducted on a double‐blind basis; however, only Csernansky 1988 and Xiang 1997 reported the methods used to ensure blinding. Weber 1983 was single‐blind (rater only) and compared diazepam to standard care so was at high risk of performance bias. None of these studies described whether blinding was tested.
Incomplete outcome data
Csernansky 1988 used a total treatment and observation time of no more than six weeks but did not describe whether people left the study early. Weber 1983 was a longer trial and lost two people to follow‐up, however the authors gave reasons and we rated this trial as low risk of attrition bias. Bobruff 1981 and Xiang 1997 were also low risk as they accounted for all participants at the end of the study.
Selective reporting
The majority of data in this review originated from published reports. All trials reported expected outcomes (impact on TD symptoms). All studies reported results of all outcomes listed in the methods section, however we rated risk of reporting bias for three studies as unclear (Bobruff 1981; Csernansky 1988; Weber 1983) because we have had no opportunity to see protocols of these trials to compare the outcomes reported in the full publications with what was measured during the conduct of the trial.
Other potential sources of bias
Only Csernansky 1988 was at high risk of bias because participants with TD were extracted post‐hoc from a larger study. The other studies had unclear or low risk. All studies had very small sample sizes. One study used a cross‐over design (Weber 1983).
Effects of interventions
See: Summary of findings for the main comparison Benzodiazepines compared with placebo for antipsychotic‐induced tardive dyskinesia; Summary of findings 2 Benzodiazepines compared with phenobarbital (as active placebo) for antipsychotic‐induced tardive dyskinesia
1. Comparison 1. Benzodiazepines versus placebo/treatment as usual
1.1 Tardive dyskinesia symptoms: no clinically important improvement (greater than 50% improvement on any tardive dyskinesia scale)
The overall results of no clinically important improvement in TD symptoms found no benefit of benzodiazepines against placebo or no treatment after five to 10 weeks' treatment (very low quality evidence, 2 RCTs, 32 people, RR 1.12, 95% CI 0.60 to 2.09, I2 = 14%, Analysis 1.1).
1.2 Tardive dyskinesia symptoms: not any improvement
For the outcome not any improvement, we found no difference between benzodiazepines and placebo or no treatment after five to 10 weeks' treatment (2 trials, 32 people, RR 1.49, 95% CI 0.33 to 6.74, I2 = 0%, Analysis 1.2).
1.3 Tardive dyskinesia symptoms: deterioration
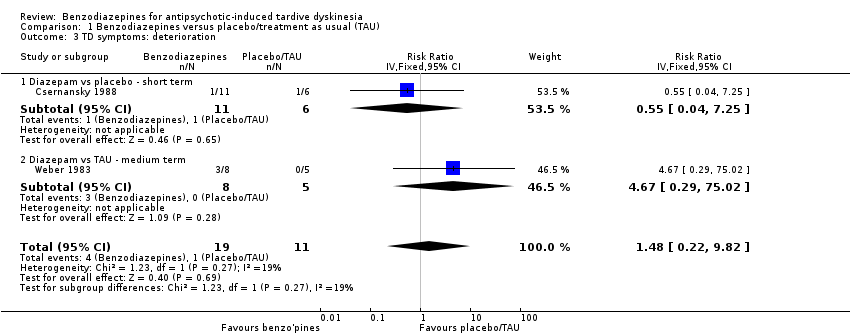
For deterioration of TD symptoms, there was no difference between benzodiazepines and placebo or no treatment after five to 10 weeks' treatment (very low quality evidence, 2 trials, 30 people, RR 1.48, 95% CI 0.22 to 9.82, I2 = 19%, Analysis 1.3).
1.4 Tardive dyskinesia symptoms: mean tardive dyskinesia score at the end of treatment
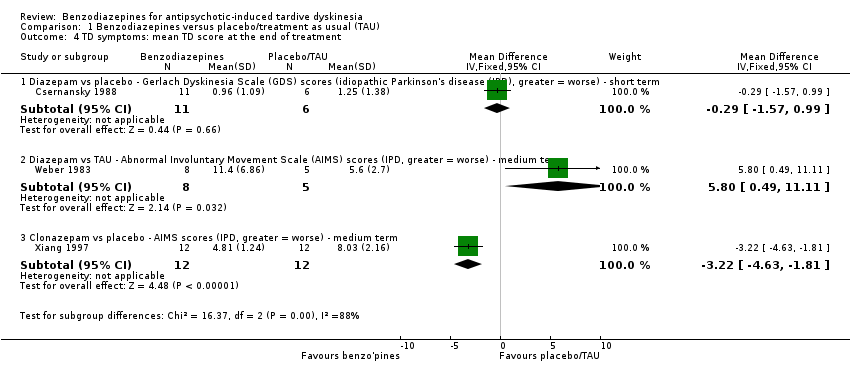
TD symptoms were also measured on the continuous AIMS and GDS scales (see Description of studies). Scores were not pooled as heterogeneity was high (I2 = 88%, P < 0.001). Csernansky 1988 found no difference between diazepam and placebo groups after five weeks' treatment (1 RCT, 17 people, MD ‐0.29, 95% CI ‐1.57 to 0.99, Analysis 1.4). However, Weber 1983 found a benefit for treatment as usual (TAU) compared with diazepam after 10 weeks' treatment (1 RCT, 13 people, MD 5.80, 95% CI 0.49 to 11.11, Analysis 1.4), and Xiang 1997 found a beneficial effect of clonazepam after eight weeks' treatment compared with placebo (1 RCT, 24 people, MD ‐3.22, 95% CI ‐4.63 to ‐1.81, Analysis 1.4).
1.5 Mental state: mean score at the end of treatment (BPRS, low = better)
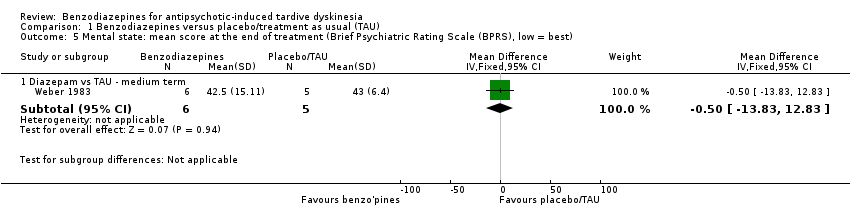
Weber 1983 reported on mental state changes (using the sum of five BPRS factors) and noted no difference between diazepam and no treatment after 10 weeks' treatment (1 RCT, 11 people, MD ‐0.50, 95% CI ‐13.83 to 12.83).
1.6 Leaving the study early
Weber 1983 reported participants leaving the study early: one person was discharged from hospital, and another continued diazepam rather than the cross‐over drug. The other three studies did not report the loss of any participants. We found no differences between groups after five to 10 weeks' treatment (very low quality evidence, 3 RCTs, 56 people, RR 2.73, 95% CI 0.15 to 48.04, Analysis 1.6).
1.7 Other issues
1.7.1 Missing outcomes
We found no data on adverse effects as they were not reported in any of the included studies.
We identified no studies that reported on hospital and service utilisation outcomes, economic outcomes, social confidence, social inclusion, social networks, personalised quality of life, behaviour, or cognitive state.
1.7.2 Subgroup analysis
a. Clinical stage: recent‐onset tardive dyskinesia
It was impossible to evaluate whether participants with recent‐onset TD responded differently to participants with more established problems, since no trial reported data for groups with different durations of TD that could be extracted for separate analyses.
b. Duration of follow‐up
There was no clear change in relation to duration of follow‐up between groups.
1.7.3 Heterogeneity
We stratified outcomes by duration of treatment (as specified in Types of outcome measures) and by intervention subtypes, i) different benzodiazepines, and ii) different control groups (placebo or no treatment), but also synthesised data when statistical heterogeneity was not high (I2 > 50%) (as specified in Assessment of heterogeneity). Data were nevertheless homogeneous for studies over time and for different intervention subtypes, except for TD symptoms: mean endpoint scores, where there was statistical heterogeneity detected (I2 = 88%, P < 0.001, see Analysis 1.4).
1.7.4 Sensitivity analyses
a. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. As all studies were stated to be randomised, we did not undertake this sensitivity analysis.
b. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings when we used our assumption compared with completer data only. Using completer only data for no clinically important improvement in TD symptoms, we found no significant difference between benzodiazepines and placebo or no treatment with no substantial alteration to the direction of effect or the precision of the effect estimates (2 RCTs, 30 participants, RR 1.08, 95% CI 0.57 to 2.05, I2 = 7%, analysis not shown).
c. Risk of bias
When excluding one trial that we judged to be at high risk of bias across one or more of the domains (Weber 1983), there was no substantial alteration to the direction of effect or the precision of the effect estimates (1 RCT, 17 participants, RR 0.73, 95% CI 0.24 to 2.23, analysis not shown).
d. Imputed values
We planned to undertake a sensitivity analysis to assess the effects of including data from cluster randomised trials where we used imputed values for ICC in calculating the design effect. No cluster randomised trials were included.
e. Fixed and random effects
We also synthesised data for the primary outcome using a random‐effects model. This did not alter the significance of the results (2 RCTs, 32 participants, RR 1.18, 95% CI 0.59 to 2.33, analysis not shown).
2. Comparison 2. Benzodiazepines versus other compounds
2.1 Tardive dyskinesia symptoms: no clinically important improvement (greater than 50% improvement on any tardive dyskinesia scale) ‐ short term
One trial found that clonazepam was more beneficial than phenobarbital (as active placebo) after two weeks' treatment (very low quality evidence, 1 RCT, 21 people, RR 0.44, 95% CI 0.20 to 0.96, Analysis 2.1).
2.2 Tardive dyskinesia symptoms: not any improvement ‐ short term
For the outcome not any improvement, one trial found no difference between clonazepam and phenobarbital (as active placebo) after two weeks' treatment (1 RCT, 21 people, RR 0.36, 95% CI 0.02 to 8.03, Analysis 2.2).
2.3 Adverse effects
One trial reported that 10/10 participants experienced adverse effects in the clonazepam group and 7/11 in the phenobarbital group, and the trial found no difference between groups after two weeks' treatment (1 RCT, 21 people, RR 1.53, 95% CI 0.97 to 2.41, Analysis 2.3).
2.4 Leaving the study early
One trial reported that no participants left the study early. Consequently, this outcome could not be estimated (see Analysis 2.4).
2.5 Other issues
2.5.1 Missing outcomes
No studies reported on mental state, hospital and service utilisation outcomes, economic outcomes, social confidence, social inclusion, social networks, personalised quality of life, behaviour, or cognitive state.
2.5.2 Subgroup analysis
Only one study was identified for this comparison and no subgroup analyses were conducted.
2.5.3 Sensitivity analysis
Only one study was identified for this comparison and no sensitivity analyses were conducted.
Discussion
Summary of main results
1. The search
This area of research does not seem to be active. The 2017 update has identified additional data, but all trials predated 2000. This could be due to a decreased concern with TD, less emergence of the problem in research‐active communities because of more thoughtful use of antipsychotic drugs, or loss of faith in benzodiazepines as a potential treatment.
2. Few data
Fewer than 100 people have been involved in placebo‐controlled trials of benzodiazepines for TD with reported outcome measures. It is possible that real and important effects have not been highlighted because of the necessarily wide CIs of the findings. Many outcomes were not measured (see Overall completeness and applicability of evidence). We may have been overambitious in hoping for some of these outcomes in TD trials, but simple reporting of adverse events and social impact/quality of life does not seem too demanding and is of interest to patients and carers.
3. Comparison 1. Benzodiazepines versus placebo/treatment as usual
3.1 Tardive dyskinesia symptoms
Results from this review indicated that whether the outcome was no clinically important improvement, not improved at all, or deterioration, there was no compelling evidence that benzodiazepines affect TD between six weeks and six months. However, since evidence was of very low quality (see summary of findings Table for the main comparison), we have very little confidence in the effect estimates and CIs; the true effects are likely to be substantially different.
In one study, there was some suggestion that the AMS score of people taking clonazepam decreased after eight weeks (Xiang 1997). We were not entirely sure that the AMS was a published scale (and therefore meeting our minimal entry criteria), and were unsure what the decline of four points means in terms of clinical signs and symptoms (see Figure 5). In any event, this finding was taken from one trial involving only 24 people, so should be viewed with caution and not as reliable evidence that clonazepam helps people with TD.

In another study, measures on the AIMS scale indicated that no treatment reduced TD symptom scores compared with diazepam (Weber 1983). Again, these results could not be taken as reliable evidence and should be interpreted with caution, not least because only 13 people were randomised.
3.2 Adverse effects
There was no suggestion that use of benzodiazepines was unacceptable for people with TD, but none of the studies comparing benzodiazepines with placebo or TAU specifically reported on adverse effects.
3.3 Mental state
There was very low quality evidence of no difference between diazepam and no treatment on mental state from one study; therefore, we have very little confidence in the effect estimate and CIs; the true effect is likely to be substantially different.
3.4 Leaving the study early
Two studies reported no events and one study reported that two participants left the intervention group compared with none in the control group. There was very low quality evidence of no difference between diazepam and no treatment on leaving the study early; therefore, we have very little confidence in the effect estimate and CIs; the true effect is likely to be substantially different.
3.5 Social confidence, social inclusion, social networks, or personalised quality of life
This group of outcomes was selected as being of importance to patients for the 2016 review update following a service user consultation. No studies were identified that reported on any of these outcomes.
4. Comparison 2. Benzodiazepines versus active other compounds
4.1 Tardive dyskinesia symptoms
Results from one study indicated that significantly more participants taking diazepam than taking phenobarbital improved to a clinically important level; however, evidence was of very low quality making our confidence in the effect estimate and CIs very low; the true effect is likely to be substantially different.
4.2 Adverse effects
Results from one study indicated no difference between benzodiazepines and phenobarbital on adverse events; however, evidence was of very low quality making our confidence in the effect estimate and CIs very low; the true effect is likely to be substantially different.
4.3 Mental state
No study reported on mental state.
4.4 Leaving the study early
One study reported no events; the effect for this outcome could not be estimated.
4.5 Social confidence, social inclusion, social networks, or personalised quality of life
This group of outcomes was selected as being of importance to patients for the 2016 review update following a service user consultation. No studies were identified that reported on any of these outcomes.
Overall completeness and applicability of evidence
1. Completeness
It is disappointing that so few high‐quality data could be extracted from relevant literature. While the evidence from small open studies is crucial in the exploratory phase of psychopharmacological research, confirmatory well‐designed, conducted, and reported randomised studies are required to assess the efficacy of an intervention properly. Benzodiazepines for TD are clearly not a very active area of research.
All four included studies were no more than pilot studies. The total numbers included in these studies was only 15 to 24 (total 75 people). Due to their small size, they cannot really be expected to fully answer any questions about the effects of benzodiazepines for people with TD. Nevertheless, these studies illustrated that trials addressing the effects of benzodiazepines for TD are possible.
There were no data on the patient‐designated important outcomes of social confidence, social inclusion, social networks, or personalised quality of life, neither were there data on hospital and service utilisation outcomes, economic outcomes, behaviour, or cognitive response. Further, there were no data on adverse events for the comparison with placebo or no treatment. It is possible that if used in the medium to long term that benzodiazepines could well have effects in these areas. Benzodiazepines can be sedating, induce tolerance, dependence, and a withdrawal syndrome (O'Brien 2005).
2. Applicability
Trials were a mixture of hospital based and outpatient, and studied people who would be recognisable in everyday care. Benzodiazepines are readily accessible and most outcomes understandable in terms of clinical practice. Should benzodiazepines have had important effects, the findings may well have been applicable.
Quality of the evidence
Overall, the quality of the evidence in this review was very low. This means that we have very little confidence in the effect estimates, and the true effects are likely to be substantially different from the estimates of the effect. We found three main reasons for our low confidence.
-
Poor study methodology and reporting of methods (see Figure 2) resulting in downgrading evidence for risk of bias. Allocation concealment was not described, generation of the sequence was not explicit, studies were not clearly blinded, and we were unsure if data were incomplete or selectively reported or if other biases were operating.
-
Very small sample sizes resulting in downgrading evidence for imprecision. The largest trial in this review randomised only 24 people. A trial of this size is unable to detect subtle, yet important differences due to benzodiazepines with any confidence. To detect a 20% difference between groups, probably about 150 people are needed in each arm of the study (alpha 0.05, beta 0.8).
-
Wide CIs (often due to low event rates) that included appreciable benefit or harm for the intervention as well as no effect, resulting in downgrading evidence for imprecision.
See summary of findings Table for the main comparison and summary of findings Table 2 for full details.
The small trial size, along with the poor reporting of trials, would be associated with an exaggeration of effect of the experimental treatment if an effect had been detected (Jűni 2001).
Potential biases in the review process
1. Missing studies
We made every effort to identify relevant trials. However, these studies were all small and it is likely that we did not identify other studies of limited power. It is likely that such studies would also not be in favour of the benzodiazepine group. If they had been so, it is more likely that they would have been published in accessible literature. We do not, however, think it likely that we have failed to identify large relevant studies.
2. Missing data
We excluded two studies (Godwin‐Austen 1971; Thaker 1990) that provided no usable data (see Excluded studies). We contacted the author of one study who replied to confirm that there were no usable data. We could not find up‐to‐date contact details for authors of the other study. We found it very unlikely that we would receive a reply from authors regarding research conducted so many years ago; therefore, these studies were excluded.
3. Introducing bias
This review has now been updated several times. Review authors have tried to be balanced in the appraisal of the evidence but could have inadvertently introduced bias. We welcome comments or criticisms. New methods and innovations now make it possible to report data where, in the past, we could not report data at all or had to report data in a different way. We believe the 'Summary of findings' tables are a valuable innovation but problematic to those not 'blind' to the outcome data. It is possible to 'select' significant findings for presentation in this table. We have tried to decrease the chance of doing this by asking a new review author (HB) to select outcomes relevant for this table before becoming familiar with the data.
Agreements and disagreements with other studies or reviews
The only other relevant quantitative review we know of are the previous Cochrane Reviews (Bhoopathi 2006; Soares‐Weiser 1999; Walker 2003). This update expanded this review, but did not substantially change the findings or the conclusions. Findings from other similar reviews suggest that TD, rather than these interventions, are no longer a focus of research activity.

Message from one of the participants of the public and patient involvement consultation of service user perspectives on tardive dyskinesia research.
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Study flow diagram.


Comparison 1 Benzodiazepines versus placebo/treatment as usual (TAU), Outcome 1 Tardive dyskinesia (TD) symptoms: no clinically important improvement (> 50% improvement on any TD scale).

Comparison 1 Benzodiazepines versus placebo/treatment as usual (TAU), Outcome 2 TD symptoms: not any improvement.
Comparison 1 Benzodiazepines versus placebo/treatment as usual (TAU), Outcome 3 TD symptoms: deterioration.
Comparison 1 Benzodiazepines versus placebo/treatment as usual (TAU), Outcome 4 TD symptoms: mean TD score at the end of treatment.
Comparison 1 Benzodiazepines versus placebo/treatment as usual (TAU), Outcome 5 Mental state: mean score at the end of treatment (Brief Psychiatric Rating Scale (BPRS), low = best).

Comparison 1 Benzodiazepines versus placebo/treatment as usual (TAU), Outcome 6 Leaving the study early.

Comparison 2 Benzodiazepines vs other compounds, Outcome 1 Tardive dyskinesia (TD) symptoms: no clinically important improvement (> 50% improvement on any TD scale) ‐ short term.
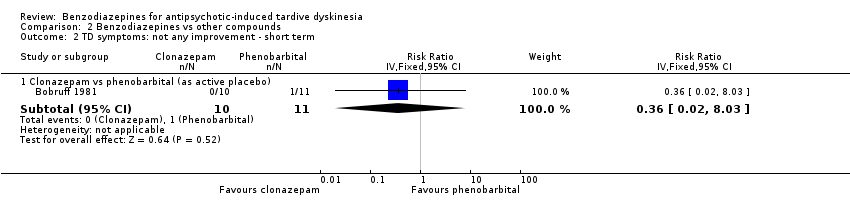
Comparison 2 Benzodiazepines vs other compounds, Outcome 2 TD symptoms: not any improvement ‐ short term.
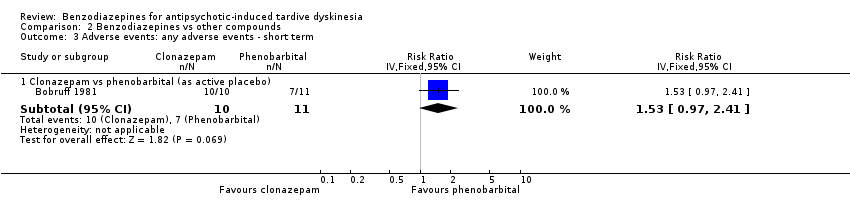
Comparison 2 Benzodiazepines vs other compounds, Outcome 3 Adverse events: any adverse events ‐ short term.

Comparison 2 Benzodiazepines vs other compounds, Outcome 4 Leaving the study early ‐ short term.
| Study tag | Participants | Comparison | Review |
| Antipsychotic‐induced akathisia | Clonazepam vs placebo | ||
| Benztropine vs propranolol | Anticholinergics for neuroleptic‐induced acute akathisia; Central action beta‐blockers versus placebo for neuroleptic‐induced acute akathisia. | ||
| Benzodiazepines vs artane (trihexyphenidyl hydrochloride). | Benzodiazepines for neuroleptic‐induced acute akathisia; Anticholinergics for neuroleptic‐induced acute akathisia. | ||
| Antipsychotic‐induced tardive dyskinesia | Naltrexone vs placebo | Miscellaneous treatments for neuroleptic‐induced tardive dyskinesia. | |
| Naltrexone + clonazepam vs clonazepam + placebo |
| Methods | Allocation: randomised. |
| Participants | Diagnosis: serious mental illness treated by antipsychotic drugs for a protracted period. Tardive dyskinesia.a |
| Interventions | 1. Clonazepam 6‐12 mg oral daily dose. |
| Outcomes | Tardive dyskinesia: any clinically important improvement in tardive dyskinesia, any improvement, deterioration.b |
| aThis could be diagnosed by clinical decision. If funds were permitting all participants could be screened using operational criteria, otherwise a random sample should suffice. bPrimary outcome. The same applies to the measure of primary outcome as for diagnosis. Not everyone may need to have operational criteria applied if clinical impression is proved to be accurate. | |
| n: number of participants. | |
| Benzodiazepines compared with placebo for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: psychiatric patients (mainly schizophrenia) with antipsychotic‐induced tardive dyskinesia | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Risk with placebo/no treatment | Risk with benzodiazepines | |||||
| Tardive dyskinesia: no clinically important improvement | Study population | RR 1.12 | 32 | ⊕⊝⊝⊝ | ‐ | |
| 545 per 1000 | 611 per 1000 | |||||
| Tardive dyskinesia: deterioration in symptoms | Study population | RR 1.48 | 30 | ⊕⊝⊝⊝ | ‐ | |
| 91 per 1000 | 135 per 1000 | |||||
| Adverse effect: any adverse event | None of the included studies reported on these outcomes. | |||||
| Adverse effect: no clinically significant extrapyramidal adverse effects | ||||||
| Acceptability of the treatment (measured by participants leaving the study early) | Study population | RR 2.73 | 56 | ⊕⊝⊝⊝ | ‐ | |
| 0 per 1000 | 0 per 1000 | |||||
| Social confidence, social inclusion, social networks, or personalised quality of life ‐ not reported | None of the included studies reported on this outcome. | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded one level for risk of bias: none of the studies adequately described randomisation procedure or allocation concealment, one study did not blind participants and personnel, and one study was a post hoc subgroup analysis of participants with tardive dyskinesia. 2Downgraded two levels for imprecision: small sample size, and 95% CI of effect estimate includes both appreciable benefit and appreciable harm for benzodiazepines. | ||||||
| Benzodiazepines compared with phenobarbital (as active placebo) for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: psychiatric patients (mainly schizophrenia) with antipsychotic‐induced tardive dyskinesia | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Risk with phenobarbital (active placebo) | Risk with benzodiazepines | |||||
| Tardive dyskinesia: no clinically important improvement | Study population | RR 0.44 | 21 | ⊕⊝⊝⊝ | ‐ | |
| 909 per 1000 | 400 per 1000 | |||||
| Tardive dyskinesia: deterioration in symptoms ‐ not measured | The included study did not report on this outcome. | |||||
| Adverse events: any | Study population | RR 1.53 | 21 | ⊕⊝⊝⊝ | ‐ | |
| 636 per 1000 | 974 per 1000 | |||||
| Adverse effect: extrapyramidal symptoms ‐ not reported | The included study did not report on this outcome. | |||||
| Acceptability of the treatment (measured by participants leaving the study early) | Study population | Not estimable | 21 | ⊕⊝⊝⊝ | No events were reported; no one left the study early. | |
| 0 per 1000 | 0 per 1000 | |||||
| Social confidence, social inclusion, social networks, or personalised quality of life ‐ not measured | The included study did not report on this outcome. | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded one level for risk of bias: the included study did not adequately describe randomisation procedure, allocation concealment, or blinding. 2Downgraded two levels for imprecision: only one study with a very small sample size. | ||||||
| Interventions | Reference |
| Anticholinergic medication | Soares‐Weiser 1997; Soares‐Weiser 2000; 2016 update to be published. |
| Benzodiazepines | This review. |
| Calcium channel blockers | Essali 2011; 2016 update to be published. |
| Cholinergic medication | Tammenmaa 2002; 2016 update to be published. |
| Gamma‐aminobutyric acid agonists | Alabed 2011; 2016 update to be published. |
| Miscellaneous treatments | Soares‐Weiser 2003; 2016 update to be published. |
| Neuroleptic reduction or cessation (or both) and neuroleptics | Soares‐Weiser 2006; 2016 update to be published. |
| Non‐neuroleptic catecholaminergic drugs | El‐Sayeh 2006; 2016 update to be published. |
| Vitamin E | Soares‐Weiser 2011; 2016 update to be published. |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Tardive dyskinesia (TD) symptoms: no clinically important improvement (> 50% improvement on any TD scale) Show forest plot | 2 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.60, 2.09] |
| 1.1 Diazepam vs placebo ‐ short term | 1 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.24, 2.23] |
| 1.2 Diazepam vs TAU ‐ medium term | 1 | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.5 [0.71, 3.16] |
| 2 TD symptoms: not any improvement Show forest plot | 2 | 32 | Risk Ratio (IV, Fixed, 95% CI) | 1.49 [0.33, 6.74] |
| 2.1 Diazepam vs placebo ‐ short term | 1 | 17 | Risk Ratio (IV, Fixed, 95% CI) | 0.55 [0.04, 7.25] |
| 2.2 Diazepam vs TAU ‐ medium term | 1 | 15 | Risk Ratio (IV, Fixed, 95% CI) | 2.5 [0.39, 16.05] |
| 3 TD symptoms: deterioration Show forest plot | 2 | 30 | Risk Ratio (IV, Fixed, 95% CI) | 1.48 [0.22, 9.82] |
| 3.1 Diazepam vs placebo ‐ short term | 1 | 17 | Risk Ratio (IV, Fixed, 95% CI) | 0.55 [0.04, 7.25] |
| 3.2 Diazepam vs TAU ‐ medium term | 1 | 13 | Risk Ratio (IV, Fixed, 95% CI) | 4.67 [0.29, 75.02] |
| 4 TD symptoms: mean TD score at the end of treatment Show forest plot | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 Diazepam vs placebo ‐ Gerlach Dyskinesia Scale (GDS) scores (idiopathic Parkinson's disease (IPD), greater = worse) ‐ short term | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐0.29 [‐1.57, 0.99] |
| 4.2 Diazepam vs TAU ‐ Abnormal Involuntary Movement Scale (AIMS) scores (IPD, greater = worse) ‐ medium term | 1 | 13 | Mean Difference (IV, Fixed, 95% CI) | 5.80 [0.49, 11.11] |
| 4.3 Clonazepam vs placebo ‐ AIMS scores (IPD, greater = worse) ‐ medium term | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | ‐3.22 [‐4.63, ‐1.81] |
| 5 Mental state: mean score at the end of treatment (Brief Psychiatric Rating Scale (BPRS), low = best) Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Diazepam vs TAU ‐ medium term | 1 | 11 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐13.83, 12.83] |
| 6 Leaving the study early Show forest plot | 3 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.73 [0.15, 48.04] |
| 6.1 Clonazepam vs placebo ‐ medium term | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.2 Diazepam vs placebo ‐ short term | 1 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.3 Diazepam vs TAU ‐ medium term | 1 | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.73 [0.15, 48.04] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Tardive dyskinesia (TD) symptoms: no clinically important improvement (> 50% improvement on any TD scale) ‐ short term Show forest plot | 1 | Risk Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 Clonazepam vs phenobarbital (as active placebo) | 1 | 21 | Risk Ratio (IV, Fixed, 95% CI) | 0.44 [0.20, 0.96] |
| 2 TD symptoms: not any improvement ‐ short term Show forest plot | 1 | Risk Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 Clonazepam vs phenobarbital (as active placebo) | 1 | 21 | Risk Ratio (IV, Fixed, 95% CI) | 0.36 [0.02, 8.03] |
| 3 Adverse events: any adverse events ‐ short term Show forest plot | 1 | Risk Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 Clonazepam vs phenobarbital (as active placebo) | 1 | 21 | Risk Ratio (IV, Fixed, 95% CI) | 1.53 [0.97, 2.41] |
| 4 Leaving the study early ‐ short term Show forest plot | 1 | Risk Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 Clonazepam vs phenobarbital (as active placebo) | 1 | 21 | Risk Difference (IV, Fixed, 95% CI) | 0.0 [‐0.17, 0.17] |