Opioids for neuropathic pain
Abstract
Background
This is an updated version of the original Cochrane review published in Issue 3, 2006, which included 23 trials. The use of opioids for neuropathic pain remains controversial. Studies have been small, have yielded equivocal results, and have not established the long‐term profile of benefits and risks for people with neuropathic pain.
Objectives
To reassess the efficacy and safety of opioid agonists for the treatment of neuropathic pain.
Search methods
We searched CENTRAL, on The Cochrane Library (Issue 10 of 12, 2012), MEDLINE (1966 to Oct week 3, 2012), and EMBASE (1980 to 2012, week 42) for articles in any language, and reference lists of reviews and retrieved articles. Searches were originally run in 2005, then again in 2010 and 2012.
Selection criteria
We included randomized controlled trials (RCTs) in which opioid agonists were given to treat central or peripheral neuropathic pain of any etiology. Pain was assessed using validated instruments, and adverse events were reported. We excluded studies in which drugs other than opioid agonists were combined with opioids or opioids were administered epidurally or intrathecally.
Data collection and analysis
Two review authors independently extracted data and included demographic variables, diagnoses, interventions, efficacy, and adverse effects.
Main results
Thirty‐one trials met our inclusion criteria, studying 10 different opioids: 23 studies from the original 2006 review and eight additional studies from this updated review.
Seventeen studies (392 participants with neuropathic pain, average 22 participants per study) provided efficacy data for acute exposure to opioids over less than 24 hours. Sixteen reported pain outcomes, with contradictory results; 8/16 reported less pain with opioids than placebo, 2/16 reported that some but not all participants benefited, 5/16 reported no difference, and 1/16 reported equivocal results. Six studies with about 170 participants indicated that mean pain scores with opioid were about 15/100 points less than placebo.
Fourteen studies (845 participants, average 60 participants per study) were of intermediate duration lasting 12 weeks or less; most studies lasted less than six weeks. Most studies used imputation methods for participant withdrawal known to be associated with considerable bias; none used a method known not to be associated with bias. The evidence, therefore, derives from studies predominantly with features likely to overestimate treatment effects, i.e. small size, short duration, and potentially inadequate handling of dropouts. All demonstrated opioid efficacy for spontaneous neuropathic pain. Meta‐analysis demonstrated at least 33% pain relief in 57% of participants receiving an opioid versus 34% of those receiving placebo. The overall point estimate of risk difference was 0.25 (95% confidence interval (CI) 0.13 to 0.37, P < 0.0001), translating to a number needed to treat for an additional beneficial outcome (NNTB) of 4.0 (95% CI 2.7 to 7.7). When the number of participants achieving at least 50% pain relief was analyzed, the overall point estimate of risk difference between opioids (47%) and placebo (30%) was 0.17 (95% CI 0.02 to 0.33, P = 0.03), translating to an NNTB of 5.9 (3.0 to 50.0). In the updated review, opioids did not demonstrate improvement in many aspects of emotional or physical functioning, as measured by various validated questionnaires. Constipation was the most common adverse event (34% opioid versus 9% placebo: number needed to treat for an additional harmful outcome (NNTH) 4.0; 95% CI 3.0 to 5.6), followed by drowsiness (29% opioid versus 14% placebo: NNTH 7.1; 95% CI 4.0 to 33.3), nausea (27% opioid versus 9% placebo: NNTH 6.3; 95% CI 4.0 to 12.5), dizziness (22% opioid versus 8% placebo: NNTH 7.1; 95% CI 5.6 to 10.0), and vomiting (12% opioid versus 4% placebo: NNTH 12.5; 95% CI 6.7 to 100.0). More participants withdrew from opioid treatment due to adverse events (13%) than from placebo (4%) (NNTH 12.5; 95% CI 8.3 to 25.0). Conversely, more participants receiving placebo withdrew due to lack of efficacy (12%) versus (2%) receiving opioids (NNTH ‐11.1; 95% CI ‐20.0 to ‐8.3).
Authors' conclusions
Since the last version of this review, new studies were found providing additional information. Data were reanalyzed but the results did not alter any of our previously published conclusions. Short‐term studies provide only equivocal evidence regarding the efficacy of opioids in reducing the intensity of neuropathic pain. Intermediate‐term studies demonstrated significant efficacy of opioids over placebo, but these results are likely to be subject to significant bias because of small size, short duration, and potentially inadequate handling of dropouts. Analgesic efficacy of opioids in chronic neuropathic pain is subject to considerable uncertainty. Reported adverse events of opioids were common but not life‐threatening. Further randomized controlled trials are needed to establish unbiased estimates of long‐term efficacy, safety (including addiction potential), and effects on quality of life.
PICOs
Plain language summary
Opioids for neuropathic pain
Neuropathic pain is pain caused by nerve damage. It is often difficult to diagnose and treat. The use of opioids (strong pain killers such as morphine) to treat neuropathic pain is controversial owing to concerns about addiction and beliefs that this type of pain does not always respond well to opioids. The review looked at short‐term studies lasting less than a day and intermediate‐term trials lasting from several days to 12 weeks. The 31 studies found involved 1237 people with neuropathic pain; most studies were small.
Short‐term studies produced mixed results, with just over half indicating that opioids might be better than a placebo. While intermediate‐term studies all indicated that opioids were better than placebo, most studies were small, most were short, and none used methods known to be unbiased. All these features are likely to make effects of opioids look better in clinical trials than they are in clinical practice. We cannot say whether opioids are better than placebo for neuropathic pain over the long term. Side effects such as constipation, nausea, dizziness, and drowsiness were common, but not life‐threatening.
Authors' conclusions
Background
This review is an update of a previously published review in the Cochrane Database of Systematic Reviews (Issue 3, 2006) on 'Opioids for neuropathic pain'.
Description of the condition
The percentage of people suffering from neuropathic pain is unknown, but is estimated to be as high as 7% to 8% (Bouhassira 2008; Torrance 2006) in developed nations. Estimates of the prevalence of chronic pain (of which neuropathic pain is a subset) suggest that around 20% of both developed and undeveloped nations' populations are affected (Breivik 2004; Breivik 2006). Neuropathic pain may result from a large variety of insults to the peripheral or central somatosensory nervous system, including trauma, inflammation, ischemia, and metabolic and neoplastic disorders. Common examples of peripheral neuropathic pain include diabetic neuropathy and postsurgical neuralgia. Central neuropathic pain includes central post‐stroke pain, pain in multiple sclerosis, and pain after spinal cord injury. The main clinical characteristics of neuropathic pain are continuous or intermittent spontaneous pain, typically described as burning, aching, or shooting in quality, and abnormal sensitivity of the painful site to normally innocuous stimuli such as light touch by garments, running water, or even wind (allodynia) (Baron 2010; Maier 2010). Neuropathic pain, like many other forms of chronic pain, often has negative effects on quality of life (Jensen 2007; Meyer‐Rosberg 2001). Pharmacotherapy for neuropathic pain has generally involved the use of antidepressants or anticonvulsants, but even with the current generation of these drugs, effective analgesia is achieved in less than half of this population (Dworkin 2010; Finnerup 2010; O'Connor 2009; Sindrup 1999).
Description of the intervention
Opioids are the most effective broad‐spectrum analgesics available and are considered the cornerstone of therapy for moderate‐to‐severe acute pain or pain of similar intensity due to life‐threatening illnesses, but their long‐term use in non‐cancer pain, of which neuropathic pain is a component, is controversial. In the United States, the therapeutic use of opioids in general has risen significantly over the last decade (Manchikanti 2008). Despite this, the safety and efficacy of the different opioids in the treatment of neuropathic pain have yet to be established. Clinical trials assessing the efficacy of opioids for reducing neuropathic pain have been reported for more than 25 years, yet great variability in trial design in terms of the type of neuropathic pain syndrome treated, the type of opioid administered, and the duration of treatment has yielded contradictory results. Studies that have suggested efficacy have used small study populations, raising questions about the validity of the results.
How the intervention might work
Opioids provide analgesia by binding to opioid receptors of the mu and kappa class and blocking the release of neurotransmitters such as substance P. Opioid receptors are expressed both centrally and peripherally during the inflammatory response in injured tissue.
Why it is important to do this review
There is a lack of definitive evidence regarding the efficacy of opioids in reducing neuropathic pain in general, and central neuropathic pain in particular. Equally, there are concerns about tolerability of opioids and the potential for abuse, addiction, hormonal abnormalities, dysfunction of the immune system, and, in some cases, paradoxical hyperalgesia with long‐term use (Rhodin 2010; Seghal 2012; Tompkin 2011; Vallejo 2004). Therefore, we conducted a systematic review of published randomized controlled trials (RCTs).
Objectives
We attempted to answer two questions:
1) What is the efficacy of opioid agonists in relieving neuropathic pain?
2) What is the nature and incidence or severity of adverse effects caused by opioid agonists in people with neuropathic pain?
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs) in this review if opioid agonists (but not partial agonists or agonist‐antagonists) were given to treat central or peripheral neuropathic pain of any etiology. Studies with pain intensity as the primary or secondary outcome were included. Non‐randomized studies and case reports were excluded, as were retrieved trials that presented insufficient data to allow assessment of the outcomes of interest or study quality.
Types of participants
We included men and women of all ages and races or ethnicities. We excluded studies in which participants with both neuropathic and other types of pain (e.g. nociceptive) were enrolled and responses of the two groups were not presented separately.
Types of interventions
We included studies in which one or more opioid agonists or different doses of the same opioid agonist were compared with placebo, each other, or another class of medication used for neuropathic pain (e.g. antidepressants). We included studies in which drugs were administered by any of the following routes: oral, rectal, transdermal, intravenous, intramuscular, or subcutaneous.
We excluded studies in which: drugs other than opioid agonists were combined with opioids (e.g. codeine with acetaminophen); opioids were administered epidurally or intrathecally, as the epidural route is usually reserved for postoperative/labor pain, and while the intrathecal route is used in neuropathic pain (usually via an implantable pump) such therapy is typically classified as neuro‐modulation rather than analgesia; or if tramadol or tapentadol were used as the active drug, because, although both interact to some degree with opioid receptors, they are not regarded as pure opioid agonists. The efficacy of tramadol in relieving neuropathic pain has been reviewed elsewhere (Duehmke 2006).
Types of outcome measures
We included participant‐reported measure(s) of pain intensity or pain relief using validated methods.
Primary outcomes
In our updated review the primary outcomes we sought were the proportion of participants reporting at least 33% pain reduction from baseline or 50% or more pain reduction from baseline. The selection of these outcomes, as opposed to the primary outcomes in our original review (mean pain intensity difference or mean pain relief) was based on the observation that pain relief tends to be bimodal, rendering mean values less useful. A greater than or equal to 33% (or ≥ 30%) pain reduction from baseline was based upon analyses demonstrating that such a reduction was required for people with chronic pain to perceive a clinically meaningful change in pain intensity (Farrar 2001). More recent evidence suggests that at least 50% pain relief is clinically significant, because high levels of pain relief are strongly associated with improved fatigue, sleep, depression, work ability and quality of life (Moore 2010a). While such data are rarely reported in older studies, we anticipated that those studies found in our updated search would report them.
Where studies did not report numbers of participants with at least 33% or 50% improvement, but reported numbers of participants reporting certain categories of global impression of change, e.g. "much improved", we translated these categories to equate to either at least 33% pain reduction from baseline or at least 50% pain reduction from baseline (Dworkin 2008).
Secondary outcomes
We extracted data on the following secondary outcomes:
-
Pain intensity or pain intensity difference or pain relief using a visual analog scale (VAS) or numerical rating scale (NRS).
-
Outcomes based on pain questionnaires and quality of life (QoL) measurement instruments, including those recommended as core chronic pain outcome domains (Dworkin 2008) (Multidimensional Pain Inventory and Brief Pain Inventory interference scales, Beck Depression Inventory and Profile of Mood States).
-
Incidence of adverse events during treatment with opioid or control (intermediate‐term studies only).
-
Participant dropouts due to adverse events (intermediate‐term studies only).
-
Participant dropouts due to lack of efficacy (intermediate‐term studies only).
We normalized pain intensity data assessed by means other than a 0 ‐ 100 VAS to such a scale. To do so we either multiplied the original scale employed by an appropriate factor (e.g. by ten if the original scale was a 0 ‐ 10 scale) or by assigning values on a 0 ‐ 100 scale that corresponded to choices on the original assessment scale. For example, if a participant was offered a five‐point scale, selection of the third point was scored as 50 on a 0 ‐ 100 scale (0 = no pain, 1 = 25, 2 = 50, 3 = 75, 4 = 100).
Search methods for identification of studies
This search was run for the original review in June 2005 and subsequent searches were run on the 16th of August, 2010. Finally, further searches employing different search strategies were run on the 24th of October 2012. The new search strategies were developed because the older strategies produced an impractical number of references for only two years of literature.
Electronic searches
We searched the following databases:
-
CENTRAL, on The Cochrane Library (Issue 10 of 12, 2012)
-
MEDLINE (1966 to Oct week 3, 2012)
-
EMBASE (1980 to to 2012 week 42 )
We combined search terms for RCTs with terms for opioids and terms for neuropathic pain. Our original search strategies can be found in Appendix 1; Appendix 2; and Appendix 3. Our updated search strategies (2010 to 2012) can be found in Appendix 4; Appendix 5; and Appendix 6.
There was no language restriction.
Searching other resources
We scanned the reference lists of reviews and retrieved articles. We did not consider abstracts or unpublished reports in this update, but intend to include them in future updates.
Data collection and analysis
Selection of studies
We determined eligibility by reading the abstract of each study identified by the search. We eliminated studies that clearly did not satisfy our inclusion criteria, and obtained full copies of the remaining studies. Two review authors read these studies independently and reached agreement by discussion. The studies were not anonymized in any way before assessment.
Data extraction and management
Two review authors extracted and agreed on data, using a standard form, before entry into Review Manager 5 (RevMan). Data extracted included information on study design and duration, methods, interventions, pain outcomes, adverse events, diagnoses, participant inclusion and exclusion criteria, numbers enrolled and completing the study, and functional assessments. We resolved discrepancies in extracted data by discussion prior to their inclusion in the analyses.
Analyses focused on differences in pain intensity, pain relief, and the incidence and severity of adverse events. When necessary and possible we normalized all data to a 0 ‐ 100 mm VAS. We made no attempt to convert surrogate outcomes (e.g. amount of rescue medication used) to a VAS, although we did equate certain global evaluations to either 33% or 50% pain reduction. For studies in which surrogate outcomes were the only results available, we describe them as such. We extracted the number of participants experiencing adverse events from trials in which they were asked about or observed for specific adverse effects such as constipation, also noting withdrawals if described.
Assessment of risk of bias in included studies
In our original review, we graded included studies for methodological quality using the Oxford Quality Scale (Jadad 1996). In our updated review, we instead assessed 'Risk of bias' for both the original included studies and those included from the updated search (see Assessment of risk of bias in included studies). Two review authors independently assessed the risk of bias of all included studies. The review authors made critical assessments for each of the following domains: sequence generation (randomization), allocation concealment, blinding, incomplete outcome data, and selective outcome reporting. The review author judgment for each domain was entered into a 'Risk of bias' table, with answers 'low risk', 'high risk' or 'unclear risk' (indicating either lack of information or uncertainty over the potential for bias).
Measures of treatment effect
In contrast to our original review, we applied a random‐effects model to combine data, in part because of the heterogeneity apparent in many of the analyses. We are aware of the possible limitation of using a random‐effects model for meta‐analysis in case of non‐normal distribution of intervention effect data; however, using a fixed‐effect model for this purpose may be less appropriate since we cannot assume to know the direction of the effect.
Dichotomous data
Discrete events such as numbers of participants reporting 33% pain relief or better, or 50% pain relief or better, or the number of participants reporting adverse events were used to calculate the risk difference using Review Manager 5 software. When a statistically significant risk difference existed between interventions, we derived the number needed to treat for an additional beneficial outcome (NNTB) or for an additional harmful outcome (NNTH) (Cook 1995). Additionally, dichotomous outcomes are presented in terms of both raw numbers and percentages of participants in each study arm benefiting from therapy or suffering adverse events.
Continuous data
We undertook meta‐analyses when comparable data were available from continuous outcomes. Comparisons between opioids and active control or placebo groups were made separately for pain relief, pain intensity post‐intervention, and intensity of a specific adverse event, using weighted mean differences (WMDs).
Unit of analysis issues
We split the control treatment arm between active treatment arms in a single study if the active treatment arms were not combined for analysis.
Dealing with missing data
We did not contact authors for original data unless data were missing or unclear. If, despite attempts to contact study authors, participant data were missing, analyses were based on participant populations in which outcomes were reported. Discrepancies between the number of participants enrolled and the number of participants in whom outcomes were reported are noted in the Characteristics of included studies table. Where studies reported statistics based on intention‐to‐treat (ITT) or modified ITT populations, we performed available case analyses. The ITT population consisted of participants who were randomized, took the assigned study medication, and provided at least one post‐baseline assessment.
Assessment of heterogeneity
We evaluated heterogeneity between and within trials using both the Chi² test and the I² test. The Chi² test assesses whether observed differences in results are compatible with chance alone. A low P value (or a large Chi² statistic relative to its degrees of freedom) provides evidence of heterogeneity of treatment effects (variation in effect estimates beyond chance). The Chi² test has low power in estimating heterogeneity in the common situation where few trials are analyzed or where included trials have small sample sizes. Although a statistically significant result may indicate a problem with heterogeneity, a non‐significant result is not necessarily evidence of lack of heterogeneity. Methods developed for quantifying inconsistency across studies that move the focus away from testing whether heterogeneity is present to assessing its impact on the meta‐analysis include the I² statistic. I² = [(Q ‐df)/Q] x 100%, where Q is the Chi² statistic and df is its degrees of freedom (Deeks 2011; Higgins 2003). The I² statistic describes the percentage of the variability in effect estimates that is due to heterogeneity rather than sampling error (chance). A value greater than 50% may be considered substantial heterogeneity (Deeks 2011). We also assessed heterogeneity by visually studying forest plots.
Assessment of reporting biases
We made no attempt to assess reporting bias.
Data synthesis
We used the random‐effects model by DerSimonian and Laird (Deeks 2011) for meta‐analysis, using Review Manager 5.
Subgroup analysis and investigation of heterogeneity
Where possible we performed subgroup analysis based on:
-
peripheral versus central pain
-
spontaneous versus evoked pain
Sensitivity analysis
For our updated review, we decided to perform sensitivity analyses by eliminating:
-
cross‐over studies
-
studies with fewer than 10 participants in an intervention arm or phase
Results
Description of studies
Results of the search
Our 2005 literature search yielded 3823 citations (CENTRAL, 945; MEDLINE, 1531; EMBASE 1347), of which we selected 46 potentially relevant studies for retrieval. The literature search covering 2005 to 16 August 2010 yielded an additional 2409 citations (CENTRAL, 370; MEDLINE, 1025; EMBASE, 1014) of which we selected 19 studies for retrieval. Finally, the 2012 search yielded 2296 citations (CENTRAL, 157; MEDLINE, 1213; EMBASE, 926) of which we selected nine for retrieval (Figure 1).
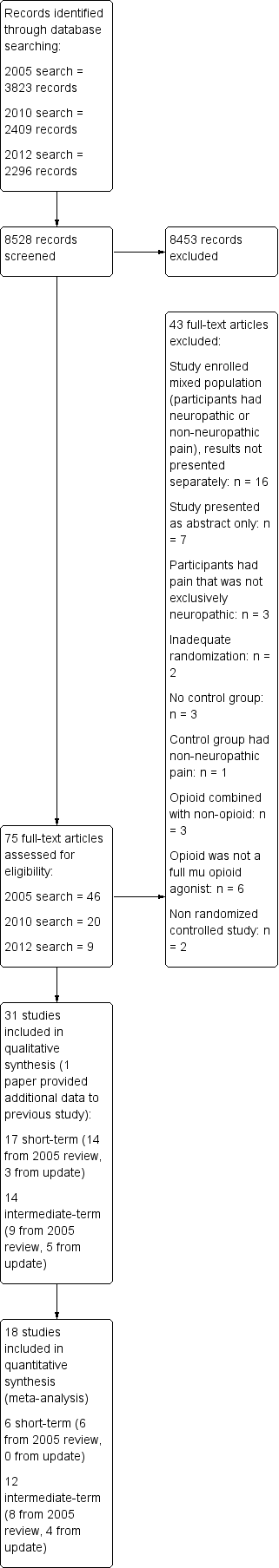
Study flow diagram.
Included studies
We divided the trials into two categories according to study duration. There is no definition per se of what constitutes a short‐term or an intermediate‐term trial. Short‐term trials, intuitively, were those that employed a single dose or intravenous infusion intervention. We labeled other trials as 'intermediate‐term' because we did not consider trial duration to be sufficiently long to make firm conclusions about chronic administration of opioids. In total, our updated review included 17 short‐term studies (three from the 2005 ‐ 2012 searches) and 14 intermediate‐term studies (five from the 2005 ‐ 2012 search).
Eight of the 28 retrieved articles from the updated search (2010, 2012) met the inclusion criteria and provided data on an additional 510 participants with neuropathic pain who were treated with opioids. Three studies were short‐term trials: Juarez‐Pichardo 2009 (N = 27); Simpson 2007 (N = 79); and Wallace 2006 (N = 32). The other five studies were intermediate‐term trials (Frank 2008; Hanna 2008; Khoromi 2007; Wu 2008; Zin 2010), in which opioids were administered orally over periods of between 35 and 84 days (median = 49 days). Numbers of participants per treatment group ranged from 29 to 169 (median = 50).
Twenty‐three of the 46 articles from our 2005 search met the inclusion criteria and provided data on 727 participants with neuropathic pain who were treated with opioids.
The first group consisted of 14 short‐term trials (treated as 16 comparisons) (Arner 1988; Attal 2002; Dellemijn 1997; Eide 1994; Eide 1995; Jadad 1992; Jorum 2003; Kupers 1991 central; Kupers 1991 peripheral; Leung 2001; Max 1988; Max 1995; Rabben 1999; Rowbotham 1991; Wu 2002 phantom limb; Wu 2002 stump) in which opioids were administered mostly as brief intravenous infusions and outcomes were measured for less than 24 hours. The number of participants in each of these studies was small (median = 13; range, 7 to 53). We subanalyzed reported outcomes from two of the trials. In one study people with both peripheral and central pain were included and the results reported separately (Kupers 1991 central; Kupers 1991 peripheral). In another study changes in phantom limb pain and stump pain were reported separately (Wu 2002 phantom limb; Wu 2002 stump).
The second group of studies consisted of nine intermediate‐term trials (Gilron 2005; Gimbel 2003; Harke 2001; Huse 2001; Morley 2003; Raja 2002; Rowbotham 2003; Watson 1998; Watson 2003) in which opioids were administered orally over longer periods of between eight and 70 days (median = 28 days), generally to larger numbers of participants (median = 57; range, 12 to 159).
Excluded studies
Three controlled trials (Benedetti 1998; Kalman 2002; Maier 2002) failed to meet one or more of the inclusion criteria in our original review. First, an RCT conducted over seven days (Maier 2002) compared morphine with placebo in a mixed group of participants with various neuropathic and nociceptive pain syndromes. The authors reported that "the number of responders was significantly higher in patients with neuropathic than with nociceptive pain". However, efficacy and adverse event data were not presented separately for the different types of pain. Second, a short‐term, placebo‐controlled trial (Kalman 2002) showed that only four of 14 participants who had multiple sclerosis and central neuropathic pain were categorized as 'responders' to intravenous morphine. The study was non‐randomized and single blinded. Third, in an RCT (Benedetti 1998), five different doses of buprenorphine (0.033 to 0.166 mg) were administered randomly to 21 participants with post‐thoracotomy neuropathic pain one month after surgery, with reduction of pain by 50% in each person. However, buprenorphine is a partial mu receptor agonist, with different pharmacological properties to members of the full µ opioid agonist class.
Twenty studies were excluded from the updated search (2005 ‐ 2012). Three trials included participants in whom pain could not be attributed entirely to neuropathic origin: in two of them participants with acute herpes zoster were enrolled (Guo 2007;Dworkin 2009); and the third consisted of participants with low back pain (Kalso 2007). The potential cause of pain in these participants can be nociceptive, neuropathic or both, and the effect of opioid treatment on the two types of pain was not reported separately. Similarly, six other trials (Ashburn 2011; Cruciani 2012; Nicholson 2006a; Nicholson 2006b; Webster 2010; Weil 2009) were excluded because enrolled participants had mixed pain syndromes (neuropathic or nociceptive) and results were not presented independently. Two trials included participants with neuropathic pain who were treated with opioids but were not RCTs (Arita 2008; Gatti 2009). Two studies had no control group (Mordarski 2009; Yao 2012). In one study the control group did not have neuropathic pain (Niesters 2011). Finally, six trials were published in an abstract form only (Buynak 2009; Hale 2009; Oh 2012; Podolsky 2009; Varrassi 2011; Webster 2011).
Risk of bias in included studies
Our original review used the Oxford Quality Scale to assess the quality of each included study. In the updated review we replaced this scale with the 'Risk of bias' tool, applying it both to new studies and to those from the original review. A summary of 'Risk of bias' assessments can be found in Figure 2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Fifteen of the 31 studies described methods for randomization adequately so that they could be assigned a low risk of bias for sequence generation. Ten of the 31 described methods of allocation concealment sufficiently that they could be assigned a low risk of bias. In both cases, the remaining studies did not provide enough information for us to draw any conclusion regarding risk of bias. Also in both cases, the majority of intermediate‐term studies had a low risk of bias, whereas a minority of short‐term studies described methods adequately. Among the intermediate‐term studies, low risk of bias was seen in similar numbers between parallel and cross‐over studies.
Blinding
Only five of the 17 short‐term studies described methods of blinding sufficiently to be assigned a low risk of bias. Conversely, 10 of the 14 intermediate‐term studies were assigned a low risk of bias, with similar numbers between parallel and cross‐over studies. All other studies were assigned an unclear risk of bias due to inadequate information.
Incomplete outcome data
Given their nature, the majority of short‐term studies (13/17) had a low risk of attrition bias. Among the intermediate‐term studies only six of 14 were assigned a low risk of bias, with one study (Rowbotham 2003) assigned a high risk. In this study 12/43 participants in the high‐dose levorphanol arm and 3/38 in the low‐dose arm withdrew from the study due to adverse events. All other studies were assigned an unclear risk: Frank 2008 reported both available case analysis and per protocol analysis, but a greater number of subjects withdrew due to side effects while assigned an opioid; in Gilron 2005 withdrawals (16/57) were evenly distributed among groups, but reasons for withdrawal not fully described; Gimbel 2003 and Hanna 2008 imputed data using last observation carried forward (LOCF); Huse 2001 made no mention of how missing data were imputed; in Morley 2003 only study completers were analyzed, six participants withdrew from the high‐dose phase due to severe nausea ‐ three while taking placebo, three while taking methadone ‐ while number of withdrawals were small, group sizes were also small. Finally, Raja 2002 employed an ITT analysis. For participants who did not complete a treatment period, the last three available pain ratings were used. However, the number of participants who did not complete the opioid phase was not reported.
Selective reporting
The vast majority of both short‐ and intermediate‐term studies reported data on all of the outcomes described in their Methods sections. While study protocols were not available for any of the reported trials, and it is therefore possible that certain outcomes were measured but not reported, the widespread reporting of pain intensity and pain relief values leads us to believe that reporting was complete for most studies.
Other potential sources of bias
Treatment group size was an issue, particularly in short‐term studies. Numbers enrolled in each study ranged from 7 to 79 in short‐term trials, with five studies enrolling fewer than 10 participants (Arner 1988; Eide 1994; Eide 1995; Jadad 1992; Max 1995) and a further five enrolling fewer than 20 participants (Attal 2002; Jorum 2003; Kupers 1991 central; Leung 2001; Rowbotham 1991). In intermediate‐term studies, enrolled arms (parallel studies) or phases (cross‐over studies) ranged in size from 12 to 169 participants. Amongst the intermediate‐term studies, the mean number of participants enrolled in each arm in parallel studies was 60, whereas the mean total enrolment size in cross‐over studies was 52. Studies with small group sizes may overestimate efficacy (Moore 1998; Nuesch 2010).
Evidence from trials in people with arthritis shows that studies lasting less than eight weeks overestimate the effect of treatment (Moore 2010b); the same may be true in studies of neuropathic pain, given that typically both are chronic conditions. Of the 14 intermediate‐term studies, nine had treatment phases that lasted less than eight weeks (Frank 2008; Gilron 2005; Gimbel 2003; Harke 2001; Huse 2001; Morley 2003; Watson 1998; Watson 2003; Zin 2010), and only one study was conducted over 12 weeks (Hanna 2008). The five parallel studies had a mean duration of 6.4 weeks, whereas the nine cross‐over studies had a mean duration of 5.7 weeks.
Effects of interventions
Short‐term studies
Our updated search added two cross‐over and one parallel study, which provided additional efficacy data for opioids in 125 participants with neuropathic pain. None of the three studies presented data in a format that we were able to add to our short‐term study meta‐analyses. In total, 17 RCTs provided efficacy data for acute exposure to opioids in 392 participants with neuropathic pain. In the three new studies, drugs were administered buccally (Simpson 2007) and intravenously (Juarez‐Pichardo 2009; Wallace 2006). In total, drugs were administered intravenously in 14 trials, orally in one (Max 1988), intramuscularly in one (Rabben 1999) and buccally in one (Simpson 2007). The duration of treatment varied from seconds (i.e. a single intramuscular injection) to eight hours, but was less than one hour in 10 trials. The tested drug was morphine in seven trials, alfentanil in four, fentanyl in two, and oxycodone, meperidine, codeine, or the investigational drug CJC‐1008 (a chemical modification of the opioid peptide dynorphin A) in one trial each. Placebo was used as a control in 14 trials. The diagnosis was specified in all trials: four trials included people with postherpetic neuralgia (PHN) only (Eide 1994; Max 1988; Rowbotham 1991; Wallace 2006); two involved people with post‐traumatic neuralgia (Jorum 2003; Max 1995); in seven, participants with mixed neuropathies were studied (Arner 1988; Dellemijn 1997; Jadad 1992; Juarez‐Pichardo 2009; Kupers 1991 central; Kupers 1991 peripheral; Leung 2001; Simpson 2007); two included people with central pain (Attal 2002; Eide 1995); one involved people with secondary (e.g. post‐traumatic) trigeminal neuropathy (Rabben 1999); and one enrolled participants with postamputation stump and phantom pain (Wu 2002 phantom limb; Wu 2002 stump). Considerable variation between studies in duration of treatment, and method of pain assessment allowed only limited quantitative synthesis of data.
Outcomes assessed
A change in spontaneous pain intensity was the primary outcome measure in 16 trials. Authors reported mixed results with respect to the analgesic efficacy of opioids for neuropathic pain in general and for specific conditions (i.e. PHN, post‐traumatic neuralgia, and central pain). Eight trials showed greater efficacy of the tested opioid versus placebo (Dellemijn 1997; Eide 1995; Jorum 2003; Leung 2001; Rowbotham 1991; Wallace 2006; Wu 2002 phantom limb; Wu 2002 stump) or another active intervention (tramadol, Juarez‐Pichardo 2009). In contrast, in five trials, researchers observed equivalent efficacy for opioids and placebo (Arner 1988; Attal 2002; Eide 1994; Max 1988; Max 1995). Two trials demonstrated partial efficacy, meaning that some participants responded to the opioid treatment while others did not (Jadad 1992; Rabben 1999). Another trial showed a reduction in the affective but not in the sensory component of pain (Kupers 1991 central; Kupers 1991 peripheral).
One study, from our updated search, assessed breakthrough pain (Simpson 2007). Fentanyl tablets or placebo were administered buccally to treat nine consecutive episodes of breakthrough pain in each of 79 participants with mixed neuropathies. Drug efficacy was measured for up to 120 minutes for each episode. This study also differed from the other short‐term studies in that participants had up to 21 days to complete the nine separate assessments. Because of the unique study design, we were not able to combine data from this study with data from any of the other short‐term studies. The study demonstrated statistically significant superiority of fentanyl buccal tablets over placebo for several outcomes, including summed pain intensity over 60 minutes, SPID60 (mean (standard error), 9.63 (0.75) versus 5.73 (0.72), respectively; P < 0.001) and proportion of breakthrough episodes with a at least 33% and at least 50% improvement in pain intensity from baseline compared with placebo from 10 minutes (9% versus 3%; P = 0.008) through two hours (66% versus 37%; P < 0.001).
Meta‐analysis
We combined data for meta‐analysis from four studies enrolling a total of 90 participants (Attal 2002; Kupers 1991 central; Kupers 1991 peripheral; Rowbotham 1991; Wu 2002 phantom limb; Wu 2002 stump) (Analysis 1.1). The result of the Chi² test for heterogeneity was 0.55 (P = 0.99), and the I² was 0%, indicating homogeneity between and within studies. The overall mean difference in the last measured pain intensity for active treatment versus placebo was ‐16 (on a 0 ‐ 100 visual analog scale (VAS)) (95% CI ‐23 to ‐9; P < 0.00001). We were able to conduct a subanalysis based on origin of neuropathic pain. Data from two trials including a total of 21 participants with central pain and from four trials involving 69 participants with peripheral neuropathic pain were subanalyzed (Analysis 1.1). For peripheral pain, the final pain intensity following opioid administration was 15 points lower than that after placebo (95% CI ‐23 to ‐7; P = 0.0002), whereas, for central pain, the difference was 18 points (95% CI ‐30 to ‐5; P = 0.006).
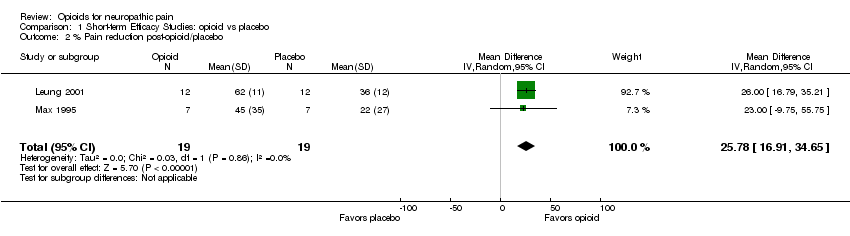
When short‐term studies were categorized according to etiology, e.g. post‐traumatic neuralgia (Jorum 2003; Max 1995), PHN (Eide 1994; Max 1988; Rowbotham 1991), the results were equivocal. One within‐study comparison (Jadad 1992) and two other between‐study comparisons (Jorum 2003 versus Max 1995 and Eide 1994 versus Rowbotham 1991) of 'high' versus 'low' opioid doses did not show an association between the opioid dose administered and analgesic efficacy. Two trials reported results in terms of percentage reduction in pain (Leung 2001; Max 1995). Meta‐analysis of these two trials demonstrated an additional 26% reduction in pain for opioid versus placebo (95% CI 17 to 35; P < 0.00001) (Analysis 1.2), although the total number of participants (N = 19) was low.
Sensitivity Analysis
Our two predetermined sensitivity analyses involved removing studies with fewer than 10 participants and removing cross‐over studies. Only one study involved in the meta‐analysis had fewer than 10 participants in a phase (Max 1995). Removing this study had minimal effect on the point estimate and no effect on statistical significance. All of the short‐term studies involved in the meta‐analyses employed a cross‐over design, therefore sensitivity analysis by study design was not possible.
Intermediate‐term studies
The results from the intermediate‐term studies are summarized in Appendix 7.
Five trials from our updated search provided data on 385 participants with neuropathic pain treated with opioids (Frank 2008; Hanna 2008; Khoromi 2007; Wu 2008; Zin 2010). The number per treatment group ranged from 29 to 169 (median 50). Three trials had a cross‐over design (Frank 2008; Khoromi 2007; Wu 2008) and two had a parallel design (Hanna 2008; Zin 2010).
When both the original and updated searches are combined, 14 trials provided data on 845 people treated with opioids. The number per treatment group ranged from 12 to 169 and the duration of treatment varied from eight days to 12 weeks. Nine trials had a cross‐over design and five had a parallel design. Five drugs were tested: morphine in six trials, oxycodone in five trials; methadone in one article comprising two trials; and levorphanol and dihydrocodeine in one trial each. Daily doses ranged from less than 5 mg to 300 mg of oral morphine equivalents, but were generally at the lower end of this range. Placebo was used as a control in all but two studies (Frank 2008; Rowbotham 2003). Five trials, in addition to opioid and placebo, included at least one arm/phase where participants received an active control or combination of interventions: carbamazepine in one trial (Harke 2001), mexiletine (Wu 2008), the tricyclic antidepressants nortriptyline and desipramine (Raja 2002; Khoromi 2007), and gabapentin (Gilron 2005).Khoromi 2007 also included a fourth arm of nortriptyline plus morphine. While many studies allowed participants to continue with any non‐opioid drug from their existing regimen, two trials specifically combined an active drug with the opioid and/or the placebo: pregabalin (Zin 2010) and gabapentin (Hanna 2008). Two trials compared different dosages of an opioid: methadone (Morley 2003) and levorphanol (Rowbotham 2003). Eight trials enrolled participants with one specific pain syndrome: diabetic neuropathy (Gimbel 2003; Hanna 2008; Watson 2003), PHN (Raja 2002; Watson 1998), chronic lumbar root pain (Khoromi 2007) and postamputation pain (Huse 2001; Wu 2008). The other studies enrolled people with neuropathic pain of diverse etiologies.
Meta‐analysis
Primary outcome ‐ proportion of participants reporting ≥ 33% pain reduction from baseline or ≥ 50% pain reduction from baseline
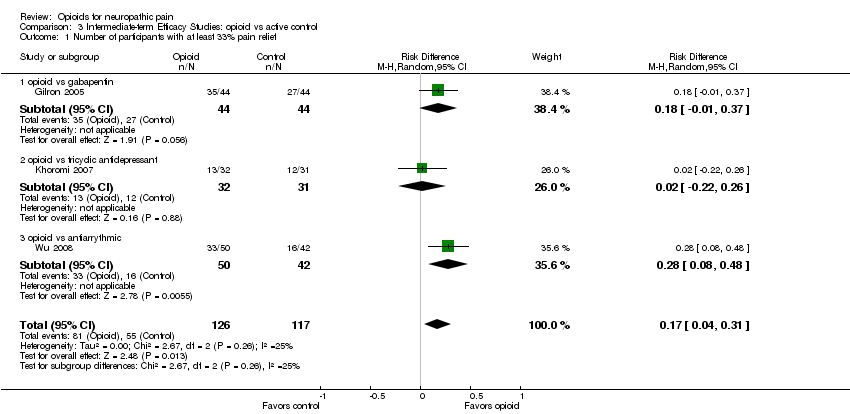
The more recent studies presented data in a format that allowed us to analyze the number of participants with at least 33% and at least 50% pain relief. For the former, 208 of 367 participants (57%) receiving an opioid achieved at least 33% relief, versus 122 of 360 participants receiving placebo (34%) (Analysis 2.1). The overall point estimate of risk difference was 0.25 (95% CI 0.13 to 0.37, P < 0.0001), translating to an NNTB of 4.0 (95% CI 2.7 to 7.7) There was significant heterogeneity (P = 0.02, I² = 63%), which was caused by the much larger risk differences (greater efficacy of opioids) in Gilron 2005 and Watson 1998 in comparison with the other studies. When these studies were removed from the analysis, heterogeneity disappeared, leaving the overall point estimate of risk difference as 0.17 (95% CI: 0.09 to 0.25, P < 0.0001), translating to an NNTB of 5.9 (95% CI 4.0 to 11.1). When number of participants achieving at least 50% pain relief was analyzed, the overall point estimate of risk difference was 0.17 (95% CI 0.02 to 0.33, P = 0.03) (Analysis 2.2), translating to an NNTB of 5.9 (3.0 to 50.0). Both the overall numbers of participants and the percentages achieving at least 50% pain relief were lower, with 72 of 154 (47%) participants receiving opioid versus 46 of 151 (30%) participants receiving placebo achieving at least 50% pain relief. One study (Zin 2010) demonstrated a tendency towards a greater number of participants in the placebo group achieving at least 50% pain relief. When this study was removed, the overall point estimate of 0.22 (95% CI: 0.09 to 0.36) in favor of those receiving an opioid, translated to an NNTB of 4.5 (95% CI 2.8 to 11.1) . A smaller number of studies assessed the number of participants with at least 33% or at least 50% pain relief when comparing opioid with an active control (Analysis 3.1; Analysis 3.2). Gilron 2005; Khoromi 2007; and Wu 2008 compared an opioid with gabapentin, a tricyclic antidepressant (nortriptyline), or an antiarrythmic (mexiletine), respectively. Only the last of these demonstrated a statistically significant difference between interventions, with morphine being superior to mexiletine when comparing the number of participants with both at least 33% pain relief (RD = 0.28, 95% CI: 0.08 to 0.48: NNTB = 3.6; 95% CI 2.1 to 12.5) and at least 50% pain relief (RD = 0.20, 95% CI: 0.01 to 0.39: NNTB = 5.0; 95% CI 2.6 to 100.0). Numbers of participants were low in this study, with 50 receiving morphine and 42 mexiletine.
Secondary outcomes
Pain intensity post‐intervention
Two studies from our updated search (Khoromi 2007; Wu 2008) added data to our 2005 analysis of pain intensity post‐intervention, comparing opioid and placebo (Analysis 2.3). Therefore, nine of the 14 studies provided data suitable for pooling. The meta‐analysis now includes 374 opioid‐treated and 351 placebo‐treated participants and shows the overall mean pain intensity to be 12 points lower in opioid‐treated participants than in those treated with placebo (95% CI ‐15 to ‐9; P < 0.00001). The addition of the two studies from our updated search (Khoromi 2007; Wu 2008) made a negligible difference to the statistical significance or point estimate. Additionally, the same two studies added data to our analysis comparing mean VAS pain scores for opioids versus active controls (Analysis 3.3). As with comparisons of the number of participants with specified percentages of pain relief, only the comparison of morphine versus mexiletine (Wu 2008) demonstrated statistically significant superiority: those receiving morphine had a 13 point lower pain intensity post‐intervention (P < 0.0001, 95% CI; ‐19 to ‐7). Again, the total number of participants was low.
Evoked pain data that we could meta‐analyze were reported in only two studies (Watson 1998; Watson 2003), with low overall numbers of participants. In both these trials oxycodone was significantly superior to placebo in reducing allodynia, categorized as 'skin pain' (Analysis 2.4). When the studies were combined statistically, participants receiving oxycodone had a 24 point lower score (95% CI: ‐34 to ‐13, P < 0.0001) on a 0 ‐ 100 VAS.
We found a dose‐dependent analgesic effect in two studies (Morley 2003; Rowbotham 2003) that included people with mixed neuropathies. In one (Morley 2003), 'low' and 'high' doses of methadone were each compared separately with placebo; the higher dose produced a greater effect than the lower dose. In the other study (Rowbotham 2003), a direct comparison showed that a higher dose of levorphanol produced a significantly greater analgesic effect than the lower dose. The use of different outcome measures in the two studies precluded the performance of a dose‐response meta‐analysis.
Quality of life and functioning
Many of the trials measured the effects of opioids on emotional and physical functioning. However, because of the use of multiple measurement tools and differences in the way data were presented, only very limited meta‐analysis with low participant numbers was possible. Several studies compared opioid versus placebo for post‐intervention results in both the physical and mental health components of the Short Form‐36 (Frank 2008; Gilron 2005; Gimbel 2003; Khoromi 2007; Watson 2003; Zin 2010), including three studies from the updated search. The studies reported mixed results; however only two reported data in a format that enabled us to perform meta‐analysis (Gilron 2005; Khoromi 2007). From these, only the sub scale 'bodily pain' demonstrated marginal superiority of opioid versus placebo, with those receiving morphine reporting a seven‐point improvement (95% CI: 0.1 to 13, Analysis 2.5). For those studies that also reported results for bodily pain, but in a format that we could not add to our meta‐analysis, two did not show an improvement of opioid over placebo (Gimbel 2003; Zin 2010), whereas Watson 2003 did report superiority. When comparing opioid versus active control, neither Gilron 2005 nor Khoromi 2007 demonstrated significant differences for any subscales, other than Khoromi 2007 reporting an 11‐point improvement in mental health for nortriptyline versus morphine (95% CI: 1 to 21) (Analysis 3.4). Two studies comparing opioid with placebo (Gilron 2005; Gimbel 2003) demonstrated improvements in several aspects of the Brief Pain Inventory, both physical and emotional, with the greatest improvement over placebo occurring in sleep, where those receiving an opioid had a 1.7 point (on an 11‐point scale) superiority over placebo (95% CI: ‐2.4 to ‐1.1, P < 0.00001, Analysis 2.6). Emotional functioning was measured by several questionnaires, including the Beck Depression Inventory and the Profile of Mood States questionnaire. We were able to meta‐analyze data from the Beck Depression Inventory when comparing both opioid and placebo, and opioid with active controls. In both cases, meta‐analysis did not demonstrate statistical differences between interventions (Analysis 2.7; Analysis 3.5), although mean scores in all groups were in the minimal to mild depression range (Dworkin 2008). Similarly, no improvement was noted in the Profile of Mood States scores of those with mixed neuropathies treated with two different dosages of levorphanol (Rowbotham 2003) nor in the Rand Mental Health Inventory completed by people with diabetic neuropathy following oxycodone treatment (Gimbel 2003).
Adverse events and withdrawals due to adverse events or lack of efficacy
As with our 2005 review, we extracted data on the incidence of common opioid‐related adverse events from all intermediate‐term studies comparing opioids with placebo. In addition, for our updated review, we analyzed adverse events when comparing opioids with active controls and divided participant withdrawals into two categories: those due to adverse events and those due to lack of efficacy, although it could be argued that both denote failure of therapy. Finally, as noted above, we used a random‐effects model, as opposed to the fixed‐effect model employed in our original review.
All but one of the intermediate‐term studies from our updated search contributed data for meta‐analysis: Frank 2008 counted each incidence of an adverse event, rather than the number of participants reporting. From our original review, Huse 2001 reported adverse events on a VAS, precluding determination of the numbers of affected participants, and Rowbotham 2003 compared two different doses of the opioid levorphanol; consequently their data could not be combined with other studies. The new studies approximately double the available participants ('N') for each analysis, increasing our confidence in their results. Although overall point estimates remained similar, the increased overall numbers meant that three of the comparisons for opioid versus placebo changed from non‐statistically significant to significant (dizziness, drowsiness and vomiting).
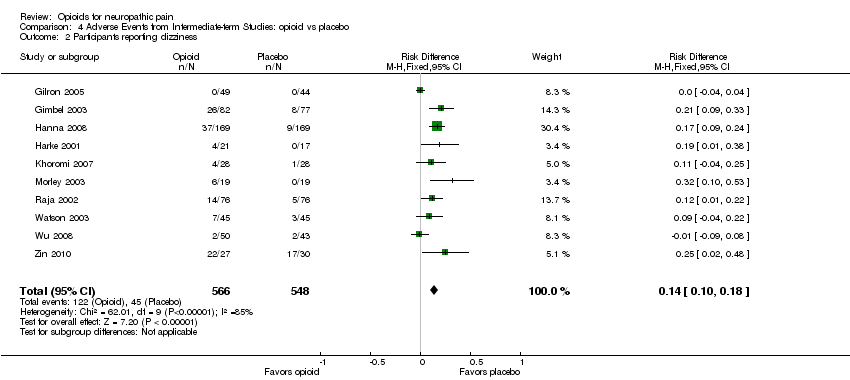
The incidence of common adverse events in both opioid and placebo groups remained similar after including the new data: constipation was the most common (34% opioid versus 9% placebo: NNTH 4.0; 95% CI 3.0 to 5.6), followed by drowsiness (29% opioid versus 14% placebo: NNTH 7.1; 95% CI 4.0 to 33.3), nausea (27% opioid versus 9% placebo: NNTH 6.3; 95% CI 4.0 to 12.5), dizziness (22% opioid versus 8% placebo: NNTH 7.1; 95% CI 5.6 to 10.0), and vomiting (12% opioid versus 4% placebo: NNTH 12.5; 95% CI 6.7 to 100.0) (Analysis 4.1; Analysis 4.2; Analysis 4.3; Analysis 4.4; Analysis 4.5). Data on cognitive impairment as well as on other adverse events were insufficient to allow calculation of the NNTH.
Our new analysis of adverse event rates for opioid versus active interventions generally demonstrated non‐statistically significant differences between treatments; however, both constipation (38% opioid versus 9% control: NNTH 3.4; 95% CI 2.6 to 4.8) and drowsiness (23% opioid versus 9% control: NNTH 7.7; 95% CI 5.0 to 16.7) occurred more frequently with opioids (Analysis 5.1; Analysis 5.3).
When opioid therapy is initiated, recipients may abandon treatment because of either adverse events or lack of efficacy. In our updated analysis, more participants withdrew from opioid treatment due to adverse events than from placebo, with 55 (13%) of 435 participants in seven studies withdrawing during opioid therapy versus 18 (4%) of 432 receiving placebo (NNTH 12.5; 95% CI 8.3 to 25.0) (Analysis 4.6). Conversely, more participants receiving placebo withdrew due to lack of efficacy, with eight of 363 (2%) participants receiving opioid withdrawing versus 42 of 360 (12%) receiving placebo (NNTH ‐11.1; 95% CI ‐20.0 to ‐8.3) (Analysis 4.7). Only one study compared withdrawal rates between opioid and an active control, nortriptyline, and did not show a statistically significant difference in rates due to either adverse events or lack of efficacy (Khoromi 2007).
Both the Chi² and I² tests for each adverse event analyzed suggested that heterogeneity existed amongst results. This may have been due to genuine differences in event rates, differences in study populations, or as a result of authors using different measurements or thresholds for reporting adverse events. In most cases, removal of one outlying study from each analysis substantially reduced heterogeneity.
Sensitivity Analysis
As with the short‐term studies, our two predetermined sensitivity analyses involved removing studies with fewer than 10 participants and removing cross‐over studies. None of the intermediate‐term studies had fewer than 10 participants in an individual arm or phase. All of the studies comparing opioid with active control in our meta‐analysis employed a cross‐over design; therefore sensitivity analysis of such studies was not possible. The majority of studies contributing data to our comparisons of opioid versus placebo also had a cross‐over design. Only five studies employed a parallel design (Gimbel 2003; Hanna 2008; Harke 2001; Rowbotham 2003; Zin 2010). Rowbotham 2003 did not contribute data to our analysis. In the few comparisons where there were sufficient numbers of both parallel and cross‐over studies, removal of cross‐over studies generally had only small effects on the point estimates of efficacy or safety, and in no cases did statistically significant overall estimates become non‐significant. Equally, the difference in overall estimates between cross‐over and parallel studies was small and did not consistently show more favorable results with cross‐over studies.
Discussion
Summary of main results
The results of this review can be divided into two categories according to the duration of included trials.
Short‐term studies
Short‐term trials demonstrated mixed results with respect to the analgesic efficacy of opioids. Our updated search did not yield any new studies suitable for statistical analysis. Although our meta‐analysis showed an overall mean difference in the last measured pain intensity for short‐term active treatment versus placebo of ‐16 points (on a 0 ‐ 100 visual analog scale (VAS)), the result should be interpreted with caution because it is based on only six of 17 studies (and only 90 of 392 participants). Thus, our conclusion regarding this category of studies has not changed from our previous meta‐analyses.
Intermediate‐term studies
Efficacy
In contrast, intermediate‐term trials demonstrated consistent opioid analgesic efficacy in reducing spontaneous neuropathic pain that was almost entirely statistically significant when results were pooled, although studies were small, mostly short, and potentially dealt inadequately with data once participants withdrew from treatment. Our updated review added data to our original analyses, increasing confidence in their findings, but also introduced new outcome analyses, i.e. number of participants with at least 33% and 50% reduction and assessments of functioning, which are considered clinically important in chronic pain (Dworkin 2008). Intermediate‐term studies are more clinically relevant than short‐term studies because they assess the benefits and risks associated with opioid treatments for weeks to months, i.e. they reflect how opioids are administered for neuropathic pain in clinical practice. This part of the meta‐analysis was based on most of the available trials and included the majority of participants. Hence, we conclude that intermediate‐term opioid treatment has a beneficial effect over placebo for spontaneous neuropathic pain as measured by both number of participants with at least 33% and at least 50% pain relief and in mean differences in post‐intervention pain intensity. Opioids did not demonstrate improvement in many aspects of emotional or physical functioning, as measured by various validated questionnaires. This raises the concern that improvements in pain relief are not accompanied by similar improvements in activities of daily living or quality of life. It should be noted, however, that our meta‐analyses of functioning included few studies, with low overall numbers of participants.
When comparing opioids with active controls, only the comparison of morphine with the rarely used antiarrythmic mexiletine demonstrated superiority of opioid (Wu 2008), with other comparisons not showing statistically significant differences between treatments. This may be due to opioids genuinely having similar efficacy to other interventions, or it may simply be a result of low participant numbers. Indirect comparisons with meta‐analyses of other treatments for neuropathic pain offer limited clarification. For example, Moore 2011 compared gabapentin with placebo for several similar efficacy outcomes. While the numbers needed to treat for an additional beneficial outcome (NNTBs) are similar for at least 50% pain relief and 33% pain relief (defined as 'moderate' relief in Moore 2011), placebo rates are much higher in the opioid analyses. Equally, participants in the gabapentin analyses often required the maximum daily dose (3600 mg), whereas in the opioid studies a larger effect was achieved by a low to moderate dose of opioid. Moreover, the dose‐dependent analgesic effects shown in two of the opioid studies (Morley 2003; Rowbotham 2003) have not been confirmed by further trials; it is therefore unclear if high doses of opioids produce a greater magnitude of pain reduction in people with neuropathic pain. Thus, we are unable to confirm the commonly held belief that opioids have no ceiling effect in this population. This may be particularly important in light of recent concerns regarding mortality risk associated with high opioid dose regimens (Gomes 2011).
Safety
Our assessment of safety did not find data related to serious adverse events, including mortality, most likely because of the relative rarity of such events, especially with low‐to‐moderate doses of opioids. Instead, we were able to analyze data related to relatively common, widely identified opioid‐induced adverse events. Not surprisingly, and in agreement with our earlier review, many of the most commonly recognized opioid side effects occurred more frequently in those treated with opioid than with placebo. Conversely, there were few statistically significant differences between opioids and active controls, which may be due to controls having similar side‐effect profiles, or to the small participant numbers for each comparison. However, constipation and drowsiness did appear to occur more commonly. In placebo‐controlled studies, 13% of participants withdrew from opioid therapy due to adverse events, and 2% due to lack of efficacy. While these percentages are not particularly high, most of the study durations were less than eight weeks; therefore numbers may increase over longer periods. Additionally, the available randomized controlled trials (RCTs) do not clearly address the issues of addiction and abuse. The absence of any report of addictive behavior or abuse in any of the intermediate‐term trials may have several explanations. It is possible that the prevalence of these behaviors is indeed low (Sullivan 2005). Alternatively, the duration of treatment in these studies may have been too short to allow such behaviors to develop. Furthermore, although not mentioned specifically as an exclusion criterion in all studies, it is reasonable to assume that the recruitment of people with active or potential abuse disorders (Dunbar 1996) into such studies would routinely be avoided. The need to further assess the risk of abuse and addiction continues to be important.
Overall completeness and applicability of evidence
Completeness
The included articles studied participants with a wide range of neuropathies, but predominately postherpetic neuralgia (PHN) and diabetic neuropathy. This reflects the distribution of neuropathic pain in the general population, with the exception that few studies included participants with back pain of neuropathic origin, which is thought to be the most common type of neuropathic pain (Torrance 2006). This may be due to the fact that it is often difficult to diagnose back pain as being purely neuropathic, with it frequently also having features of nociceptive pain. There were insufficient numbers of participants with individual neuropathies to perform a subanalysis of efficacy or safety. While our updated review added a substantial amount of data for each meta‐analysis, the overall numbers are still low for most comparisons. In particular, the inconsistency of reporting of outcomes related to functioning precludes our making firm conclusions. Many of the studies reported outcomes only on those participants completing the trial; therefore efficacy may have been over‐estimated. In those that performed an intention‐to‐treat (ITT) or modified ITT analysis, the most commonly used method of imputation was last observation carried forward (LOCF), which again may overestimate efficacy (Moore 2012). None of the studies used baseline observation carried forward (BOCF). The short duration of many intermediate‐term studies is also a potential source of bias (Moore 2010a).
Applicability
Several points deserve consideration in terms of applicability of the evidence.
First, the fact that short‐term trials, in contrast to the intermediate‐term trials, yielded inconsistent efficacy results suggests that short‐term opioid administration is unlikely to serve as a useful predictive tool when initiating a trial of opioid therapy in people with neuropathic pain.
Second, the NNTB results further confirm that opioids reduce various forms of neuropathic pain and are relatively safe, and therefore indicate that opioids at low‐to‐moderate doses are suitable for use over periods of weeks to months in the treatment of neuropathic pain. However, despite the common use of NNTB values to compare relative efficacy of different treatments, especially when head‐to‐head comparative trials are relatively scarce, their validity has been questioned for reasons such as differences in trial designs, exclusion of non‐placebo‐controlled trials, dichotomization of data, and strict and not necessarily clinically relevant cut‐off points (i.e. 50% pain relief) (Finnerup 2010).
Third, the use of a single dimension for assessment of efficacy of analgesic treatments is problematic in any form of chronic pain, which is a multi‐dimensional phenomenon. This becomes even more problematic in neuropathic pain, where even a single etiological syndrome (e.g. PHN) typically differs considerably from one patient to another in term of its clinical representation. For that reason the use of additional outcome measures rather than a single pain intensity or pain relief method have been recommended (Wittink 2005). Unfortunately, consistent improvement in specific features of neuropathic pain (e.g. evoked pain), in emotional or physical aspects of functioning, or in health‐related quality of life could not be demonstrated in the present review.
Fourth, the debate regarding the differential efficacy of opioids for central versus peripheral neuropathic pain (Ballantyne 2003; Canavero 2003; Dellemijn 1999; McQuay 1997; Nicholson 2004) has not been resolved by our study. Results of the included studies varied considerably and the meta‐analyses could not include all relevant studies. Despite limited data, the meta‐analyses showed similar opioid responsiveness for pain of central and peripheral etiologies.
Fifth, although a dose‐dependent analgesic effect was found in two studies, the dose ranges tested are still in the low‐intermediate range and do not necessarily reflect clinical practice in some countries (e.g. the USA). This, along with increasing concerns about opioid toxicity, especially at higher dose ranges (greater than the daily equivalent of 200 mg of oral morphine), does not support the use of high doses of opioids for the relief of neuropathic pain.
Lastly, this review also included a quantitative analysis of common opioid‐related adverse effects. Although the analysis is based on a relatively large number of participants with neuropathic pain, those enrolled in clinical trials may not be representative of the broader patient population seen in clinical practice. Enrolled participants have met inclusion criteria, and their willingness to enter a clinical trial suggests that they may have a higher adherence profile compared with those who are not enrolled.
Quality of the evidence
The quality of evidence improved somewhat with those articles included in our updated search. Many more of the newer studies reported outcomes considered to be clinically relevant, such as numbers of participants with at least 50% pain relief, and in a format that allowed us to perform meta‐analysis (Dworkin 2008). Additionally, the risk of bias for each domain, while low overall, was generally lower in newer studies.
Potential biases in the review process
We believe the search methodology used here to be unbiased, and the selection criteria relevant to the nature of neuropathic pain. However, two aspects of our review methodology have the potential to introduce bias to our analyses. First, we included studies of less than 12 weeks duration. While we included 'short‐term' studies purely for 'proof of concept', even amongst our 'intermediate‐term' studies only one (Hanna 2008) was conducted over 12 weeks. Studies of less than 12 weeks duration may overestimate treatment efficacy (Moore 2010a). Given the dearth of long‐term studies, we adopted a 'best available evidence' approach, and anticipate that future studies will have longer durations, allowing us to better assess the efficacy and safety of opioids administered over clinically relevant time periods.
Second, we analyzed data from cross‐over studies in the same manner as that from parallel studies. This approach may give rise to a unit‐of‐analysis error (Higgins 2011). However, as discussed (Effects of interventions) we performed a sensitivity analysis where cross‐over studies were removed from meta‐analyses and found negligible differences in estimates of effect for either efficacy or safety.
Agreements and disagreements with other studies or reviews
Most recent guidelines on the pharmacotherapy of neuropathic pain are in agreement with the results of the present review and recommend the use of opioids, typically as second‐ or third‐line treatment options (Attal 2010; Dworkin 2010; Moulin 2007; Pergolizzi 2008).
A systematic review of the evidence for pharmacological treatment of neuropathic pain (Finnerup 2010) also found that opioids have a consistent efficacy in neuropathic pain. Notably, the NNTBs for achieving meaningful pain relief in several neuropathic pain conditions (i.e. painful polyneuropathy, postherpetic neuralgia, peripheral nerve injury and mixed neuropathic pain) varied from 2.1 to 5.1 and were slightly lower than the NNTBs found in the present review. The differences in findings occurred for several reasons. First, we excluded studies with tramadol. Second, we had two additional studies in our NNTB analysis (Khoromi 2007; Zin 2010). Third, Finnerup 2010 combined results for participants with at least 33% and at least 50% pain relief, whereas we analyzed these outcomes separately. Last, and perhaps most importantly, they performed meta‐analysis separately for each neuropathic pain syndrome. We combined data as we considered numbers of participants for each syndrome to be insufficient for subanalysis.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Comparison 1 Short‐term Efficacy Studies: opioid vs placebo, Outcome 1 Pain intensity post‐opioid/placebo.
Comparison 1 Short‐term Efficacy Studies: opioid vs placebo, Outcome 2 % Pain reduction post‐opioid/placebo.

Comparison 2 Intermediate‐term Efficacy Studies: Opioid vs. Placebo, Outcome 1 Number of participants with at least 33% pain relief.

Comparison 2 Intermediate‐term Efficacy Studies: Opioid vs. Placebo, Outcome 2 Number of participants with at least 50% pain relief.

Comparison 2 Intermediate‐term Efficacy Studies: Opioid vs. Placebo, Outcome 3 Pain intensity post‐opioid/placebo.

Comparison 2 Intermediate‐term Efficacy Studies: Opioid vs. Placebo, Outcome 4 Evoked pain intensity post‐opioid/placebo.

Comparison 2 Intermediate‐term Efficacy Studies: Opioid vs. Placebo, Outcome 5 SF‐36 Health Survey.

Comparison 2 Intermediate‐term Efficacy Studies: Opioid vs. Placebo, Outcome 6 Brief Pain Inventory: Pain Interference items.

Comparison 2 Intermediate‐term Efficacy Studies: Opioid vs. Placebo, Outcome 7 Beck Depression Inventory.
Comparison 3 Intermediate‐term Efficacy Studies: opioid vs active control, Outcome 1 Number of participants with at least 33% pain relief.

Comparison 3 Intermediate‐term Efficacy Studies: opioid vs active control, Outcome 2 Number of participants with at least 50% pain relief.

Comparison 3 Intermediate‐term Efficacy Studies: opioid vs active control, Outcome 3 Pain intensity post‐opioid/active control.

Comparison 3 Intermediate‐term Efficacy Studies: opioid vs active control, Outcome 4 SF‐36 Health Survey.

Comparison 3 Intermediate‐term Efficacy Studies: opioid vs active control, Outcome 5 Beck Depression Inventory.

Comparison 4 Adverse Events from Intermediate‐term Studies: opioid vs placebo, Outcome 1 Participants reporting constipation.
Comparison 4 Adverse Events from Intermediate‐term Studies: opioid vs placebo, Outcome 2 Participants reporting dizziness.
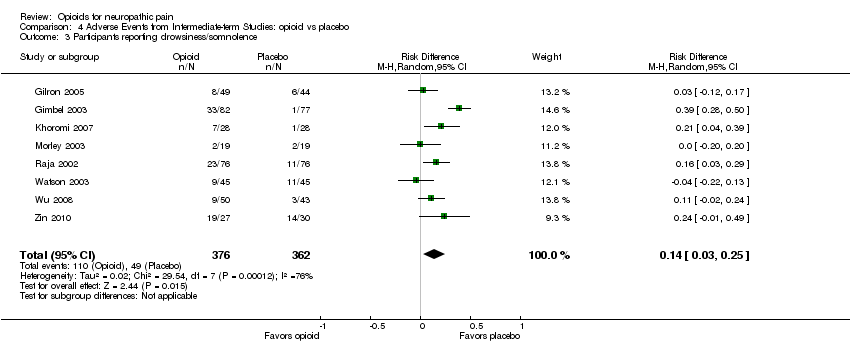
Comparison 4 Adverse Events from Intermediate‐term Studies: opioid vs placebo, Outcome 3 Participants reporting drowsiness/somnolence.

Comparison 4 Adverse Events from Intermediate‐term Studies: opioid vs placebo, Outcome 4 Participants reporting nausea.

Comparison 4 Adverse Events from Intermediate‐term Studies: opioid vs placebo, Outcome 5 Participants reporting vomiting.
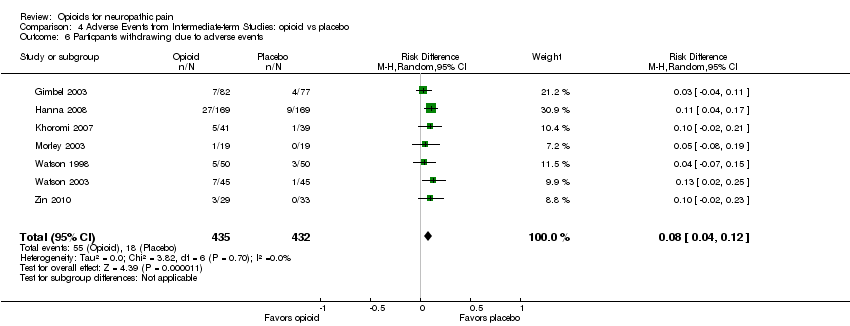
Comparison 4 Adverse Events from Intermediate‐term Studies: opioid vs placebo, Outcome 6 Particpants withdrawing due to adverse events.

Comparison 4 Adverse Events from Intermediate‐term Studies: opioid vs placebo, Outcome 7 Participants withdrawing due to lack of efficacy.

Comparison 5 Adverse Events from Intermediate‐term Studies: opioid vs active control, Outcome 1 Participants reporting constipation.
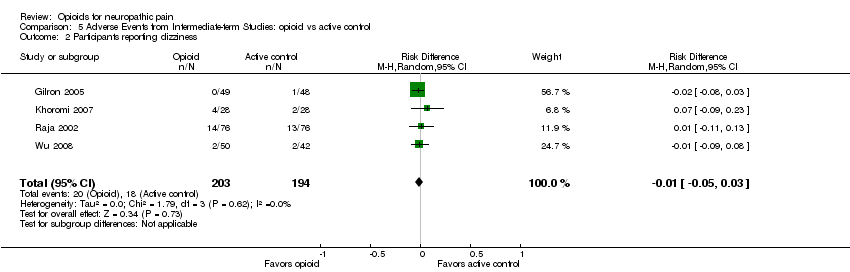
Comparison 5 Adverse Events from Intermediate‐term Studies: opioid vs active control, Outcome 2 Participants reporting dizziness.
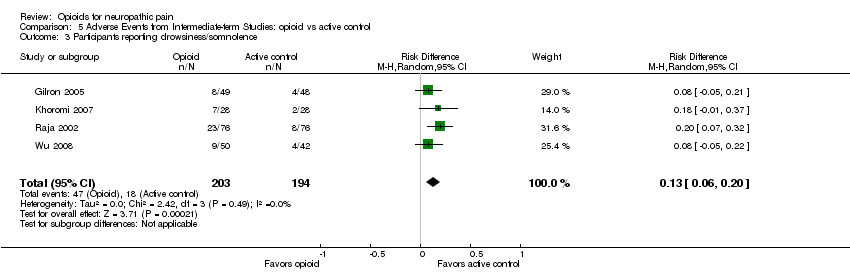
Comparison 5 Adverse Events from Intermediate‐term Studies: opioid vs active control, Outcome 3 Participants reporting drowsiness/somnolence.
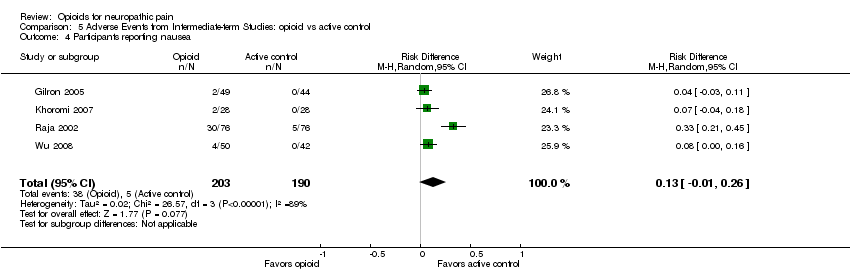
Comparison 5 Adverse Events from Intermediate‐term Studies: opioid vs active control, Outcome 4 Participants reporting nausea.

Comparison 5 Adverse Events from Intermediate‐term Studies: opioid vs active control, Outcome 5 Participants reporting vomiting.
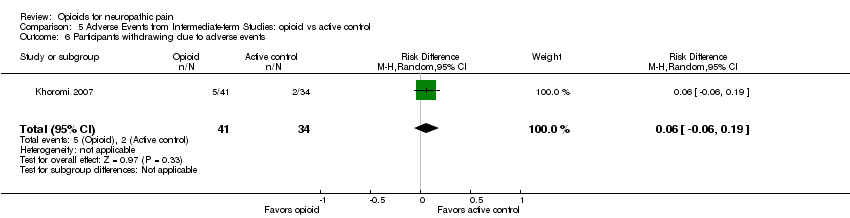
Comparison 5 Adverse Events from Intermediate‐term Studies: opioid vs active control, Outcome 6 Participants withdrawing due to adverse events.
Comparison 5 Adverse Events from Intermediate‐term Studies: opioid vs active control, Outcome 7 Participants withdrawing due to lack of efficacy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Pain intensity post‐opioid/placebo Show forest plot | 6 | 180 | Mean Difference (IV, Random, 95% CI) | ‐15.81 [‐22.54, ‐9.07] |
| 1.1 Peripheral Pain | 4 | 138 | Mean Difference (IV, Random, 95% CI) | ‐15.01 [‐22.97, ‐7.06] |
| 1.2 Central Pain | 2 | 42 | Mean Difference (IV, Random, 95% CI) | ‐17.81 [‐30.48, ‐5.15] |
| 2 % Pain reduction post‐opioid/placebo Show forest plot | 2 | 38 | Mean Difference (IV, Random, 95% CI) | 25.78 [16.91, 34.65] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Number of participants with at least 33% pain relief Show forest plot | 6 | 727 | Risk Difference (M‐H, Random, 95% CI) | 0.25 [0.13, 0.37] |
| 2 Number of participants with at least 50% pain relief Show forest plot | 5 | 305 | Risk Difference (M‐H, Random, 95% CI) | 0.17 [0.02, 0.33] |
| 3 Pain intensity post‐opioid/placebo Show forest plot | 9 | 725 | Mean Difference (IV, Random, 95% CI) | ‐12.01 [‐15.40, ‐8.62] |
| 4 Evoked pain intensity post‐opioid/placebo Show forest plot | 2 | 148 | Mean Difference (IV, Random, 95% CI) | ‐23.73 [‐34.50, ‐12.96] |
| 5 SF‐36 Health Survey Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Physical functioning | 2 | 142 | Mean Difference (IV, Random, 95% CI) | 3.16 [‐5.46, 11.77] |
| 5.2 Role‐physical | 2 | 142 | Mean Difference (IV, Random, 95% CI) | 9.62 [‐7.73, 26.97] |
| 5.3 Bodily pain | 2 | 142 | Mean Difference (IV, Random, 95% CI) | 6.78 [0.08, 13.48] |
| 5.4 General health | 2 | 142 | Mean Difference (IV, Random, 95% CI) | ‐0.62 [‐8.08, 6.85] |
| 5.5 Vitality | 2 | 142 | Mean Difference (IV, Random, 95% CI) | 1.62 [‐5.82, 9.07] |
| 5.6 Social functioning | 2 | 142 | Mean Difference (IV, Random, 95% CI) | 3.40 [‐5.09, 11.88] |
| 5.7 Role‐emotional | 2 | 142 | Mean Difference (IV, Random, 95% CI) | 7.97 [‐5.06, 21.00] |
| 5.8 Mental health | 2 | 142 | Mean Difference (IV, Random, 95% CI) | 3.09 [‐3.05, 9.23] |
| 6 Brief Pain Inventory: Pain Interference items Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 General activity | 2 | 245 | Mean Difference (IV, Random, 95% CI) | ‐0.91 [‐1.67, ‐0.14] |
| 6.2 Mood | 2 | 245 | Mean Difference (IV, Random, 95% CI) | ‐0.62 [‐1.31, 0.07] |
| 6.3 Walking | 2 | 245 | Mean Difference (IV, Random, 95% CI) | ‐0.54 [‐1.28, 0.20] |
| 6.4 Normal work | 2 | 245 | Mean Difference (IV, Random, 95% CI) | ‐0.82 [‐1.59, ‐0.05] |
| 6.5 Social relations | 2 | 245 | Mean Difference (IV, Random, 95% CI) | ‐0.71 [‐1.25, ‐0.16] |
| 6.6 Sleep | 2 | 245 | Mean Difference (IV, Random, 95% CI) | ‐1.74 [‐2.42, ‐1.06] |
| 6.7 Enjoyment of life | 2 | 245 | Mean Difference (IV, Random, 95% CI) | ‐1.18 [‐1.91, ‐0.44] |
| 7 Beck Depression Inventory Show forest plot | 3 | 273 | Mean Difference (IV, Random, 95% CI) | 0.21 [‐2.29, 2.71] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Number of participants with at least 33% pain relief Show forest plot | 3 | 243 | Risk Difference (M‐H, Random, 95% CI) | 0.17 [0.04, 0.31] |
| 1.1 opioid vs gabapentin | 1 | 88 | Risk Difference (M‐H, Random, 95% CI) | 0.18 [‐0.01, 0.37] |
| 1.2 opioid vs tricyclic antidepressant | 1 | 63 | Risk Difference (M‐H, Random, 95% CI) | 0.02 [‐0.22, 0.26] |
| 1.3 opioid vs antiarrythmic | 1 | 92 | Risk Difference (M‐H, Random, 95% CI) | 0.28 [0.08, 0.48] |
| 2 Number of participants with at least 50% pain relief Show forest plot | 2 | 155 | Risk Difference (M‐H, Random, 95% CI) | 0.07 [‐0.20, 0.33] |
| 2.1 opioid vs tricyclic antidepressant | 1 | 63 | Risk Difference (M‐H, Random, 95% CI) | ‐0.07 [‐0.30, 0.15] |
| 2.2 opioid vs antiarrythmic | 1 | 92 | Risk Difference (M‐H, Random, 95% CI) | 0.20 [0.01, 0.39] |
| 3 Pain intensity post‐opioid/active control Show forest plot | 4 | 388 | Mean Difference (IV, Random, 95% CI) | ‐7.19 [‐13.13, ‐1.25] |
| 3.1 opioid vs gabapentin | 1 | 88 | Mean Difference (IV, Random, 95% CI) | ‐5.0 [‐14.40, 4.40] |
| 3.2 opioid vs tricyclic antidepressant | 2 | 208 | Mean Difference (IV, Random, 95% CI) | ‐3.30 [‐13.48, 6.89] |
| 3.3 opioid vs antiarrythmic | 1 | 92 | Mean Difference (IV, Random, 95% CI) | ‐13.0 [‐19.12, ‐6.88] |
| 4 SF‐36 Health Survey Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Physical functioning | 2 | 144 | Mean Difference (IV, Random, 95% CI) | ‐5.09 [‐13.81, 3.63] |
| 4.2 Role‐physical | 2 | 144 | Mean Difference (IV, Random, 95% CI) | ‐5.38 [‐19.05, 8.29] |
| 4.3 Bodily pain | 2 | 144 | Mean Difference (IV, Random, 95% CI) | ‐3.11 [‐9.91, 3.70] |
| 4.4 General health | 2 | 144 | Mean Difference (IV, Random, 95% CI) | ‐4.44 [‐11.75, 2.86] |
| 4.5 Vitality | 2 | 144 | Mean Difference (IV, Random, 95% CI) | ‐6.60 [‐13.63, 0.44] |
| 4.6 Social functioning | 2 | 144 | Mean Difference (IV, Random, 95% CI) | ‐6.04 [‐14.44, 2.35] |
| 4.7 Role‐emotional | 2 | 144 | Mean Difference (IV, Random, 95% CI) | ‐6.39 [‐19.37, 6.60] |
| 4.8 Mental health | 2 | 144 | Mean Difference (IV, Random, 95% CI) | ‐6.24 [‐14.06, 1.57] |
| 5 Beck Depression Inventory Show forest plot | 3 | 276 | Mean Difference (IV, Random, 95% CI) | 1.40 [‐0.38, 3.17] |
| 5.1 opioid vs gabapentin | 1 | 88 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐2.46, 3.06] |
| 5.2 opioid vs tricyclic antidepressant | 2 | 188 | Mean Difference (IV, Random, 95% CI) | 2.17 [‐0.14, 4.49] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participants reporting constipation Show forest plot | 10 | 1114 | Risk Difference (M‐H, Random, 95% CI) | 0.25 [0.18, 0.33] |
| 2 Participants reporting dizziness Show forest plot | 10 | 1114 | Risk Difference (M‐H, Fixed, 95% CI) | 0.14 [0.10, 0.18] |
| 3 Participants reporting drowsiness/somnolence Show forest plot | 8 | 738 | Risk Difference (M‐H, Random, 95% CI) | 0.14 [0.03, 0.25] |
| 4 Participants reporting nausea Show forest plot | 10 | 1114 | Risk Difference (M‐H, Random, 95% CI) | 0.16 [0.08, 0.25] |
| 5 Participants reporting vomiting Show forest plot | 7 | 813 | Risk Difference (M‐H, Random, 95% CI) | 0.08 [0.01, 0.15] |
| 6 Particpants withdrawing due to adverse events Show forest plot | 7 | 867 | Risk Difference (M‐H, Random, 95% CI) | 0.08 [0.04, 0.12] |
| 7 Participants withdrawing due to lack of efficacy Show forest plot | 5 | 723 | Risk Difference (M‐H, Random, 95% CI) | ‐0.09 [‐0.12, ‐0.05] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participants reporting constipation Show forest plot | 4 | 397 | Risk Difference (M‐H, Random, 95% CI) | 0.29 [0.21, 0.38] |
| 2 Participants reporting dizziness Show forest plot | 4 | 397 | Risk Difference (M‐H, Random, 95% CI) | ‐0.01 [‐0.05, 0.03] |
| 3 Participants reporting drowsiness/somnolence Show forest plot | 4 | 397 | Risk Difference (M‐H, Random, 95% CI) | 0.13 [0.06, 0.20] |
| 4 Participants reporting nausea Show forest plot | 4 | 393 | Risk Difference (M‐H, Random, 95% CI) | 0.13 [‐0.01, 0.26] |
| 5 Participants reporting vomiting Show forest plot | 1 | 97 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [‐0.04, 0.04] |
| 6 Participants withdrawing due to adverse events Show forest plot | 1 | 75 | Risk Difference (M‐H, Random, 95% CI) | 0.06 [‐0.06, 0.19] |
| 7 Participants withdrawing due to lack of efficacy Show forest plot | 1 | 75 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [‐0.05, 0.05] |