Esteroides inhalados y riesgo de neumonía para la enfermedad pulmonar obstructiva crónica
References
Referencias de los estudios incluidos en esta revisión
Referencias de los estudios excluidos de esta revisión
Referencias de los estudios en espera de evaluación
Referencias de los estudios en curso
Referencias adicionales
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Design: randomised, double‐blind, parallel‐group study 12‐Month treatment period Conducted at 98 research sites in the United States and Canada | |
| Participants | Participants: 797 people were randomly assigned to fluticasone/salmeterol combination (394) and salmeterol alone (403) Baseline characteristics: Male %: flut/sal 51, sal 57 Mean age (SD), years: flut/sal 65.4 (9.1), sal 65.3 (8.8) Smoking history (mean (SD) pack‐years): flut/sal 57.8 (32.7), sal 56.5 (27.8) Mean % predicted pre‐FEV1 (SD): 34.1 (11.1), 33.9 (10.6) Inclusion criteria: ≥ 40 years of age with a diagnosis of COPD (chronic bronchitis and/or emphysema), a cigarette smoking history ≥ 10 pack‐years, a prealbuterol FEV1/FVC ≤ 0.70, an FEV1 ≤ 50% of predicted normal and a documented history of at least one COPD exacerbation the year before the study that required treatment with antibiotics or oral corticosteroids and/or hospitalisation | |
| Interventions | Run‐in: Four‐week run‐in period during which participants received open‐label FSC 250/50 via DISKUS twice daily Treatments: 1. Fluticasone/salmeterol 250/50 mcg twice daily 2. Salmeterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: Concurrent use of inhaled long‐acting bronchodilators was not allowed during the study period. Oral corticosteroids and antibiotics were allowed for short‐term treatment of a COPD exacerbation | |
| Outcomes | Primary: annual rate of moderate/severe exacerbations Secondary: time to first moderate/severe exacerbation, annual rate of exacerbations requiring oral corticosteroids, predose FEV1, time to onset of each moderate/severe exacerbation, diary records of dyspnoea scores, night‐time awakenings due to COPD and use of supplemental albuterol | |
| Notes | Funding: GSK Clinicaltrials.gov: NCT00115492 Definition of pneumonia: confirmed by chest x‐ray | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatments were assigned in blocks using a centre‐based randomisation schedule. As bronchodilator response to FSC 250/50 is generally larger in individuals with COPD who demonstrate FEV1 reversibility to albuterol, assignment to blinded study medication was stratified on the basis of participants' FEV1 response to albuterol at screening to provide a similar distribution of albuterol‐responsive and non‐responsive participants in each treatment group |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (assumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Withdrawal rates were very high compared with the numbers of events for different outcomes |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported and could be included |
| Methods | Design: double‐blind, randomised, parallel‐group trial of high‐dose inhaled budesonide versus placebo Six‐month treatment period (originally intended as 12 months) Conducted at a single centre in Canada | |
| Participants | Participants: 79 people were randomly assigned to budesonide (39) and placebo (40) Male %: bud 84.6, placebo 72.5 Mean age (SD), years: bud 66 (8), placebo 66 (8) Smoking history (mean (SD) pack‐years): bud 52 (27), placebo 50 (28) Mean % predicted pre‐FEV1 (SD): bud 36 (12), placebo 37 (10) Inclusion criteria: 40 years of age or older; smokers or ex‐smokers; absence of an exacerbation in | |
| Interventions | Run‐in: All participants were assessed in a single‐blind manner with a two‐week course of oral placebo followed by two weeks of prednisone 40 mg daily. The prednisone was subsequently tapered and was discontinued completely during the third week. Those who did not respond were randomly assigned Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: Turbohaler Co‐treatment: All medications needed for the well‐being of participants were permitted, except inhaled corticosteroids other than budesonide. In cases of treatment failure, rescue medication with beta2‐agonists or systemic steroids was available | |
| Outcomes | Primary: change in FEV1 Secondary: pre‐bronchodilator and postbronchodilator FEV1 and FVC, pre‐bronchodilator six‐minute walking test, dyspnoea with exercise, quality of life questionnaires, morning and evening PEFR, symptom scores and adverse events | |
| Notes | Funding: Astra Pharma Inc Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central computer‐generated list of random numbers |
| Allocation concealment (selection bias) | Low risk | Identification of individual treatment assignments was possible only in cases of emergency by breaking the sealed envelope kept by the investigator. The envelopes had to be kept with the case record forms and had to be returned unbroken at the end of the study |
| Blinding of participants and personnel (performance bias) | Low risk | To ensure that outcomes were measured similarly in the treatment groups, both participants and investigators were blinded to the study treatment |
| Blinding of outcome assessment (detection bias) | Low risk | To ensure that outcomes were measured similarly in the treatment groups, both participants and investigators were blinded to the study treatment |
| Incomplete outcome data (attrition bias) | High risk | Uneven dropout. Much higher in placebo group (25% vs 7.7% in the ICS group) |
| Selective reporting (reporting bias) | High risk | Key outcomes missing (mortality, adverse events). No reply from study author |
| Methods | Design: randomised, double‐blind, parallel‐group, placebo‐controlled trial Three‐month treatment period Conducted at two respiratory centres in Canada | |
| Participants | Participants: 41 people were randomly assigned to fluticasone (20) and placebo (21) Male %: flut 84.6, placebo 72.5 Mean age (SD), years: flut 66 (8), placebo 66 (8) Smoking history (mean (SD) pack‐years): flut 52 (27), placebo 50 (28) Mean % predicted FEV1 (SD): flut 36 (12), placebo 37 (10) Inclusion criteria: Age > 40 and < 75 years; smoking history (> 10 pack‐years); postbronchodilator FEV1 > 25% of predicted | |
| Interventions | Run‐in: unclear duration Treatments: 1. Fluticasone 500 mcg twice daily 2. Placebo twice daily Participants were also randomly assigned to fluticasone/salmeterol combination, but this was not included in the present review because no salmeterol arm was available for comparison Inhaler device: Diskus Co‐treatment: short‐acting bronchodilators, short‐ and long‐acting anticholinergics or theophylline was allowed throughout the study. Oral corticosteroids and/or antibiotics could be given only in short courses for exacerbation treatment | |
| Outcomes | Primary: treatment difference in the numbers of CD8+ T lymphocytes and CD68+ macrophages on bronchial biopsies Secondary: numbers of neutrophils and eosinophils in bronchial biopsies | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central computer‐generated list of random numbers |
| Allocation concealment (selection bias) | Low risk | Set up by a data management/randomisation company (GEREQ, Montreal, Quebec). A procedure was established by GEREQ, which was in possession of the treatment code, to ensure that the treatment code would be broken only in accordance with the protocol and the criteria set up for unbinding of the study |
| Blinding of participants and personnel (performance bias) | Low risk | Observers and participants were blinded to drug treatment |
| Blinding of outcome assessment (detection bias) | Low risk | Observers were blinded to whether the biopsies were performed post‐treatment or pretreatment |
| Incomplete outcome data (attrition bias) | Low risk | Dropout uneven but low in both groups (5% ICS and 14% placebo) |
| Selective reporting (reporting bias) | High risk | Key outcomes not reported. No reply from study author |
| Methods | Design: double‐blind, placebo‐controlled study Three‐year treatment period Conducted at 18 hospitals in the UK | |
| Participants | Participants: 740 people were randomly assigned to fluticasone (372) and placebo (370)Baseline characteristics: Male %: flut 75.0, placebo 74.1 Mean age (SD), years: flut 63.7 (7.1), placebo 63.8 (7.1) Smoking history (mean (SD) pack‐years): flut 44 (30), placebo 44 (34) Mean % predicted FEV1 (SD): flut 50.3 (14.9), placebo 50.0 (14.9) Inclusion criteria: current or former smokers 40 to 75 years of age with nonasthmatic chronic obstructive pulmonary disease. Baseline FEV1 after bronchodilator was at least 0.8 L but less than 85% of predicted normal, and the ratio of FEV1 to forced vital capacity was less than 70%. Previous use of inhaled and oral corticosteroids was permitted | |
| Interventions | Run‐in: eight‐week runin period after withdrawal from any oral or inhaled corticosteroids Treatments: 1. Fluticasone propionate 500 mcg daily 2. Placebo twice daily Inhaler device: metered‐dose inhaler with a spacer device Co‐treatment: Nasal and ophthalmic corticosteroids, theophyllines and all other bronchodilators were allowed during the study | |
| Outcomes | Primary: decline (mL/y) in FEV1 after bronchodilator Secondary: frequency of exacerbations, changes in health status, withdrawals due to respiratory disease, morning serum cortisol concentrations and adverse events | |
| Notes | Funding: GlaxoWellcome Research and Development Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated allocation (block size of six) |
| Allocation concealment (selection bias) | Low risk | Participants were randomly assigned sequentially from a list comprising treatment numbers only |
| Blinding of participants and personnel (performance bias) | Low risk | Three‐year double‐blind phase using an identical placebo inhaler |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Very high dropout in both groups (43% and 53%) |
| Selective reporting (reporting bias) | Unclear risk | No outcomes appear to be missing, but protocol could not be located to ensure that all were reported. Author attempted contact with GSK statistician, but no data were provided in time for publication |
| Methods | Design: multi‐centre, randomised, double‐blind, parallel‐group, placebo‐controlled study 12‐Month treatment period Conducted at 196 hospitals in 25 countries | |
| Participants | Participants: 1465 people were randomly assigned to fluticasone/salmeterol combination (358), fluticasone (374), salmeterol (372) and placebo (361) Male %: flut/salm 75, flut 70, salm 70, placebo 75 Mean age (SD), years: flut/salm 62.7 (8.7), flut 63.5 (8.5), salm 63.2 (8.6), placebo 63.4 (8.6) Smoking history (mean (SD) pack‐years): flut/salm 42.0 (22.4), flut 41.5 (20.7), salm 43.7 (21.9), placebo 43.4 (22.4) Mean % predicted FEV1 (SD): flut/salm 44.8 (14.7), flut 45.0 (13.6), salm 44.3 (13.8), placebo 44.2 (13.7) Inclusion criteria: All participants had a baseline FEV1 before bronchodilation that was 25% to 70% of that predicted, an increase of less than 10% of predicted FEV1 30 minutes after inhalation of 400 mcg salbutamol and a pre‐bronchodilator FEV1/forced vital capacity (FVC) ratio of 70% or less. Participants also had a history of at least 10 pack‐years of smoking, chronic bronchitis, at least one episode of acute COPD symptom exacerbation per year in the previous 3 years and at least one exacerbation in the year immediately before trial entry that required treatment with oral corticosteroids, antibiotics or both | |
| Interventions | Run‐in: During the 2‐week run‐in, participants stopped taking regular inhaled corticosteroids or long‐acting beta2‐agonists Treatments: 1. Fluticasone/salmeterol 500/50 mcg twice daily 2. Fluticasone 500 mcg twice daily 3. Salmeterol 50 mcg twice daily 4. Placebo twice daily Inhaler device: multi‐dose dry powder inhaler (Diskus or Accuhaler) Co‐treatment: Inhaled salbutamol was used as relief medication throughout the study, and regular treatment with anticholinergics, mucolytics and theophylline was allowed. All non‐COPD medications could be continued if the dose remained constant when possible, and if their use would not be expected to affect lung function | |
| Outcomes | Primary: FEV1 at least 6 and 12 hours after study medication Secondary: pretreatment FVC and post‐treatment FEV1 and FVC, daily record card symptoms, morning and evening PEF, use of relief medication, night‐time awakenings, acute exacerbations, health‐related quality of life (SGRQ), adverse events and electrocardiology | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A randomisation schedule generated by the participant allocation for clinical trials (PACT) programme to assign participants to study treatment groups |
| Allocation concealment (selection bias) | Unclear risk | Every participating centre was supplied with a list of participant numbers (assigned to participants at their first visit) and a list of treatment numbers. Participants who satisfied the eligibility criteria were assigned the next sequential treatment number from the list |
| Blinding of participants and personnel (performance bias) | Low risk | Salmeterol and fluticasone combination, salmeterol, fluticasone and placebo were packaged in identical inhaler devices |
| Blinding of outcome assessment (detection bias) | Unclear risk | Adverse event information was obtained at each clinic visit by recording spontaneously reported complaints from participants and asking general questions about medical troubles and concomitant medications |
| Incomplete outcome data (attrition bias) | High risk | Withdrawal rates varied from 25% in the combination inhaler group to 39% in the placebo group. These numbers are substantially higher than the numbers of events reported for any of the outcomes. The unknown outcome of participant withdrawal from the study could have a major impact on the result |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. Checked with study authors |
| Methods | Design: multi‐centre, randomised, double‐blind, parallel‐group, placebo‐controlled study 12‐Month treatment period Conducted at 109 centres in 15 countries | |
| Participants | Participants: 1022 people were randomly assigned to budesonide/formoterol combination (254), budesonide (257), formoterol (255) and placebo (256) Male %: bud/form 78, bud 74, form 75, placebo 75 Mean age (range), years: bud/form 64 (42 to 86), bud 64 (41 to 85), form 63 (41 to 84), placebo 65 (43 to 85) Smoking history (mean pack‐years): bud/form 33, bud 39, form 36, placebo 30 Mean % predicted FEV1 (SD): bud/form 36 (10), bud 36 (10), form 36 (10), placebo 36 (10) Inclusion criteria: GOLD‐defined COPD (stages III and IV); ≥ 40 years; COPD symptoms > 2 years; smoking history ≥ 10 pack‐years; FEV1/FVC ≤ 70% pre‐bronchodilator; FEV1 ≤ 50% predicted; use of short‐acting beta2‐agonists as reliever medication; ≥ 1 COPD exacerbation requiring oral corticosteroids/antibiotics 2 to 12 months before first clinic visit | |
| Interventions | Run‐in: All participants received 30 mg oral prednisolone twice daily and 2 × 4.5 mg formoterol twice daily (2 weeks) Treatments: 1. Budesonide/formoterol 320/9 mcg twice daily 2. Budesonide 400 mcg twice daily 3. Formoterol 9 mcg twice daily 4. Placebo twice daily Inhaler device: Turbuhaler Co‐treatment: terbutaline 0.5 mg as needed, courses of oral corticosteroids (maximum 3 weeks per course) and antibiotics in the event of exacerbations, parenteral steroids and/or nebulised treatment (single injections/inhalations) at emergency visits | |
| Outcomes | Primary: time to first exacerbation and change in postmedication FEV1 Secondary: number of exacerbations, time to and number of oral corticosteroid–treated episodes, change in postdose FEV1, slow VC, morning and evening PEF, quality of life (SGRQ), symptoms, use of reliever medication and adverse events | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised trial (no other details—funded by AstraZeneca and presumed to adhere to usual methods) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind (assume participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | An intention‐to‐treat analysis was used but dropout was high in all groups (ranging from 29% to 44%) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the industry document were reported in full. Checked with study authors |
| Methods | Design: multi‐centre, double‐blind, placebo‐controlled, randomised, parallel‐group study Three‐year treatment period Conducted at 444 centres in 42 countries | |
| Participants | Participants: 6184 people were randomly assigned to fluticasone/salmeterol combination (1546), fluticasone (1551), salmeterol (1542) and placebo (1545) Male %: flut/salm 75, flut 75, salm 76, placebo 76 Mean age (SD), years: flut/salm 65.0 (8.3), flut 65.0 (8.4), salm 65.1 (8.2), placebo 65.0 (8.2) Smoking history (mean (SD) pack‐years): flut/salm 47.0 (26.5), flut 49.2 (28.6), salm 49.3 (27.7), placebo 48.6 (26.9) Mean % predicted FEV1 (SD): flut/salm 44.3 (12.3), flut 44.1 (12.3), salm 43.6 (12.6), placebo 44.1 (12.3) Inclusion criteria: current or former smokers with at least a 10‐pack‐year history. Eligible individuals were 40 to 80 years of age and had received a diagnosis of COPD, with a pre‐bronchodilator forced expiratory volume in one second (FEV1) of less than 60% of predicted value, an increase in FEV1 with the use of 400 mcg of albuterol of less than 10% of the predicted value for that individual and a ratio of pre‐bronchodilator FEV1 to forced vital capacity (FVC) equal to or less than 0.70 | |
| Interventions | Run‐in: Before the 2 week run‐in period, all use of corticosteroids and inhaled long‐acting bronchodilators was stopped, but participants could continue other medications for COPD Treatments: 1. Fluticasone/salmeterol 500/50 mcg twice daily 2. Fluticasone 500 mcg twice daily 3. Salmeterol 50 mcg twice daily 4. Placebo twice daily Inhaler device: multi‐dose dry powder inhaler (Diskus or Accuhaler) Co‐treatment: all COPD medications except corticosteroids and inhaled long‐acting bronchodilators | |
| Outcomes | Primary: time to death from any cause by three years Secondary: frequency of exacerbations, health status (SGRQ), postbronchodilator spirometry and adverse events | |
| Notes | Funding: GSK Clinicaltrials.gov ID: NCT00268216 Definition of pneumonia: Study authors state there was no prospective definition because the finding was unexpected NOTE: Participants previously enrolled into TRISTAN (SFCB3024) may be recruited to this trial 4 weeks after stopping their previous study medication | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random assignment with permuted blocks with stratification |
| Allocation concealment (selection bias) | Low risk | Medication was allocated with the use of three numbers as follows.
A specialist IVR system company, ClinPhone, managed this system. At the randomisation visit (visit two), the principal investigator or designee contacted the IVR system through an automated 24‐hour telephone number; upon provision of a unique personal identification number (PIN) and answers to a series of questions, the site was provided with the participant's treatment number as well as a pack number |
| Blinding of participants and personnel (performance bias) | Low risk | Neither the subject nor the investigator knew to which treatment arm a participant had been allocated. At each treatment visit, each participant was issued with a treatment pack containing DISKUS/ACCUHALER inhalers. The inhalers contained one of the four treatments (salmeterol/fluticasone propionate combination product, fluticasone propionate, salmeterol or placebo) in accordance with the randomisation schedule. The inhalers were labelled in accordance with all applicable regulatory requirements. Each treatment pack and study treatment inhaler was labelled with the protocol number; storage and dosing instructions were provided by GW Research and Development |
| Blinding of outcome assessment (detection bias) | Unclear risk | The investigator is responsible for the detection and documentation of events meeting the definition of an AE or SAE as provided in this protocol. However, no prospective definition of pneumonia is provided in the study protocol |
| Incomplete outcome data (attrition bias) | Unclear risk | Outcome all‐cause mortality: Participants who prematurely discontinue study drug will be followed up for 156 weeks from randomisation for assessment of survival. The risk of bias due to incomplete outcome data for mortality is low General Withdrawal rates were high but fairly even (with the exception of the placebo group, which had an even higher withdrawal rate; placebo 44%, LABA 36%, ICS 38% and ICS/LABA 34%). However, for participants who withdrew from the study prematurely, all data on exacerbations, health status and lung function available at the time of a participant's withdrawal from the study were included in the analysis. All efficacy analyses were performed according to the intention‐to‐treat principle |
| Selective reporting (reporting bias) | Low risk | All collected data reported. Checked with study authors |
| Methods | Design: double‐blind, double‐dummy, randomised, active‐controlled, parallel‐group study 11‐Month treatment period (48 weeks) Conducted at 76 centres in 8 countries across Europe | |
| Participants | Participants: 481 people were randomly assigned to budesonide/formoterol combination (242) and formoterol (239) Male %: bud/form 81.5, form 81.1 Mean age (SD), years: bud/form 64.1 (9.1), form 63.7 (8.8) Smoking history (mean (SD) pack‐years): bud/form 37.8 (14.6), form 39.7 (19.1) Mean % predicted FEV1 (SD): bud/form 42.3 (6.0), form 42.5 (5.9) Inclusion criteria: hospital outpatients with severe stable COPD according to the GOLD guidelines; 40 years of age with a diagnosis of symptomatic COPD for > 2 years, at least a 20‐pack‐year smoking history, a postbronchodilator FEV1 between 30% and 50% of predicted normal and at least 0.7 L absolute value and a predose FEV1/forced vital capacity (FVC) of 0.7; at least one exacerbation requiring medical intervention (oral corticosteroid and/or antibiotic treatment and/or need for a visit to an emergency department and/or hospitalisation) within 2 to 12 months before the screening visit and the need to be clinically stable for the 2 months before study entry; change in FEV1 < 12% of predicted normal value 30 minutes following inhalation of 200 mg of salbutamol pMDI | |
| Interventions | Run‐in: During the 4‐week run‐in period, all non‐permitted COPD treatments were discontinued and eligible participants were treated with combination ipratropium/salbutamol (20/100 mg, two inhalations three times daily) Treatments: 1. Budesonide/formoterol 400/12 mcg twice daily 2. Formoterol 12 mcg twice daily Inhaler device: dry powder inhaler Co‐treatment: not described | |
| Outcomes | Primary: change in predose morning FEV1 and mean rate of COPD exacerbations per participant per year Secondary: FVC, PEF, SGRQ total score, six‐minute walking test, BMI, BODE index, safety evaluations including ECG | |
| Notes | Funding: Chiesi Farmaceutici Clinicaltrials.gov ID: NCT00476099 Definition of pneumonia: not defined | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation scheme followed a balanced‐block centre‐stratified design and was prepared via a computerised system |
| Allocation concealment (selection bias) | Low risk | Participants were centrally assigned, in each centre, to one of the three treatment arms at the end of the run‐in period through an Interactive voice/web response System (IXRS) |
| Blinding of participants and personnel (performance bias) | Low risk | On each study day, participants took both active medications and matched placebo twice daily, to maintain blinding |
| Blinding of outcome assessment (detection bias) | Low risk | On each study day, participants took both active medications and matched placebo twice daily, to maintain blinding. In cases of emergency, unbinding of the treatment code was done through IXRS |
| Incomplete outcome data (attrition bias) | Low risk | 12.3% withdrew from the combination group and 14.2% from the formoterol group. Judged to be relatively low and even between groups, and for the intention‐to‐treat population, last observation carried forward was used |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the prospectively registered protocol were reported in full |
| Methods | Design: randomised, double‐blind, placebo‐controlled trial 12‐Month treatment period in total, but participants/clinicians could stop study inhalers and return to usual (prerandomisation) steroid inhalers at any point during the study. These participants remained in the study, continued completing their diary cards and were followed up Conducted at 31 general practices in East London and Essex, UK | |
| Participants | Participants: 260 people were randomly assigned to fluticasone (128) and placebo (132) Male %: flut 48, placebo 56 Mean age (SD), years: flut 67.6 (8.9), placebo 67.3 (9.0) Smoking history (mean (SD) pack‐years): flut 40.0 (24.2), placebo 38.8 (22.3) Mean % predicted FEV1 (SD): flut 53.2 (18.2), placebo 55.0 (17.1) Inclusion criteria: Investigators searched the medical record database at each practise to identify people 40 years of age and older, with a history of smoking, who had been prescribed ICS for a minimum of six months. We invited people who fulfilled these criteria to attend a recruitment interview. Individuals with lung function consistent with international guidelines for the diagnosis of COPD were invited to join the study: postbronchodilator FEV1 less than 80% predicted, FEV1/FVC ratio less than 70% and a pre‐bronchodilator to postbronchodilator change in FEV1 of less than 15%. Participants with an FEV1 greater than 15% but with a volume change of less than 200 mL were also included | |
| Interventions | Run‐in: two‐week run‐in period before randomisation, when participants stopped their regular ICS Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily Inhaler device: Accuhaler Co‐treatment: General practitioners were advised to manage exacerbations according to usual guidance with antibiotics and/or oral steroids. Decisions about stopping study inhalers and returning to usual (prerandomisation) steroid inhalers were made by the general practitioner and the participant. Participants who did return to their usual steroid inhaler after randomisation remained in the study, continued completing their diary cards and were followed up for one year | |
| Outcomes | Primary: COPD exacerbation frequency Secondary: time to first exacerbation (from diary cards and medical records), reported symptoms, peak expiratory flow rate and reliever inhaler use (from diary cards) and lung function and health=related quality of life (at follow‐up visits) | |
| Notes | Funding: The British Lung Foundation and Newham National Health Service Trust Research and Development funded the study. GlaxoSmithKline provided the study inhalers free of charge but was not involved in study design, data collection or analysis or interpretation of results Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were allocated with minimisation to intervention or control using the programme MINIM v1.3. Minimisation factors were age, smoking status, pretrial weekly dose of ICS, self reported COPD exacerbation frequency and percentage predicted FEV1 |
| Allocation concealment (selection bias) | Low risk | Inhalers were given an alphanumerical code to conceal allocation |
| Blinding of participants and personnel (performance bias) | Low risk | Study nurses and regular clinicians were blind to allocation throughout the study. Inhalers were given an alphanumerical code to conceal allocation |
| Blinding of outcome assessment (detection bias) | Low risk | Study nurses performed the measurements and were blind to allocation |
| Incomplete outcome data (attrition bias) | Low risk | Decisions about stopping study inhalers and returning to usual (prerandomisation) steroid inhalers were made by the general practitioner and the participant. Participants who did return to their usual steroid inhaler after randomisation remained in the study, continued completing their diary cards and were followed up for one year |
| Selective reporting (reporting bias) | High risk | Key outcomes were not reported (e.g. total mortality, breakdown of all and serious adverse events). No reply from study authors |
| Methods | Design: randomised, double‐blind, parallel‐group, pilot study 12‐Month treatment period Conducted at a single centre in Italy | |
| Participants | Participants: 12 people were randomly assigned to fluticasone/salmeterol combination (6) and salmeterol alone (6) Male %: flut/salm 83.3, salm 100 Age range (mean not reported), years: flut/salm 53 to 77, salm 55 to 78 Smoking history (mean (SD) pack‐years): flut/salm 40.1 (6.3), salm 43.1 (5.3) Mean % predicted FEV1 (SD): flut/salm 50 (2.0), salm 48 (5.6) Inclusion criteria: basal FEV1 < 80% predicted normal value, but > 800 mL; FEV1/FVC ratio < 70% predicted; FEV1 change of < 12% as a percentage of predicted normal | |
| Interventions | Run‐in: Patients entered a two‐week run‐in period during which they assumed their regular treatment with theophylline and salbutamol as required Treatments: 1. Fluticasone/salmeterol 250/50 mcg twice daily 2. Salmeterol 50 mcg twice daily Participants were also randomly assigned to placebo (6), but this group was not included in the current review because no group received fluticasone monotherapy Inhaler device: Diskus Co‐treatment: theophylline and salbutamol as required | |
| Outcomes | Unclear which outcome was primary Exacerbations per year, FEV1, morning PEF, daily symptom scores, use of rescue medication and adverse events | |
| Notes | Funding: unclear Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | At the end of the run‐in, eligible participants will be randomly assigned to receive one of the three double‐blind treatments (no other details and does not appear to be industry funded) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Assigned to receive one of the three double‐blind treatments, all via Diskus |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | No dropout |
| Selective reporting (reporting bias) | High risk | Several key outcomes not reported (mortality, adverse events). Difficulty contacting study authors |
| Methods | Design: two replicate double‐blind parallel‐group trials 12‐Month treatment period Study one was conducted at 167 centres in 15 countries (Argentina, Australia, Canada, Chile, Estonia, Germany, Italy, Mexico, Netherlands, Peru, Phillipines, South Africa, Sweden, the United Kingdom and the United States). Study two was conducted at 183 centres in 15 countries | |
| Participants | Participants: 1622 people were randomly assigned in study one and 1633 in study two to vilanterol 25 (818), fluticasone/vilanterol combination 50/25 (820), 100/25 (806) and 200/25 (811) Male %: Vil 58.0, F/Vil 50/25 58.0, F/Vil 100/25 56.2, F/Vil 200/25 57.6 Mean age, years: Vil 63.6, F/Vil 50/25 63.7, F/Vil 100/25 63.8, F/Vil 200/25 63.7 Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1: Vil 45.2, F/Vil 50/25 45.4, F/Vil 100/25 46.1, F/Vil 200/25 45.2 Inclusion criteria: Eligible patients were 40 years of age or older and had a history of COPD, a smoking history of 10 or more pack‐years, a ratio of forced expiratory volume in one second (FEV1) to forced vital capacity of 0.70 or less after bronchodilators (and an FEV1 of 70% or less of predicted) and a documented history of one or more moderate or severe disease exacerbations in the year before screening | |
| Interventions | Run‐in: four weeks during which participants received open‐label combination fluticasone propionate (250) and salmeterol (50) twice daily to establish adherence to treatment and a stable baseline Treatments: 1. Fluticasone furoate/vilanterol 50/25 mcg daily 2. Fluticasone furoate/vilanterol 100/25 mcg daily 3. Fluticasone furoate/vilanterol 200/25 mcg daily 4. Vilanterol 25 daily Inhaler device: dry powder inhaler Co‐treatment: Appendix listing the exclusion criteria and the drugs that were and were not permissible for the study duration could not be located | |
| Outcomes | Primary: annual rate of moderate and severe exacerbations Secondary: time to first moderate or severe exacerbation; annual rate of exacerbations requiring oral corticosteroids; predose AM FEV1 | |
| Notes | Funding: GSK Clinicaltrials.gov ID: NCT01009463 and NCT01017952 Definition of pneumonia: confirmed by chest x‐ray | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The central randomisation schedule was generated by the GSK statistics group, which used a validated computerised system (RandAll; GSK, London, UK) |
| Allocation concealment (selection bias) | Unclear risk | The statistician entered the parameters for the randomisation but was masked to treatment assignment and did not have access to the master randomisation schedule until the study was unmasked at database lock. The Registration and Medication Ordering System (RAMOS; GSK, London, UK) was used to register and randomly assign participants and assign drugs |
| Blinding of participants and personnel (performance bias) | Low risk | Participants and investigators were masked to allocation, and the Ellipta dry powder inhalers were identical in appearance. Every effort was made to ensure that the statistics and programming department of GSK was not unmasked to any treatment allocations ahead of the database freeze |
| Blinding of outcome assessment (detection bias) | Low risk | Participants and investigators were masked to allocation, and the Ellipta dry powder inhalers were identical in appearance. Every effort was made to ensure that the statistics and programming department of GSK was not unmasked to any treatment allocations ahead of the database freeze |
| Incomplete outcome data (attrition bias) | Low risk | Dropout quite high but even between groups (ranging from 23% to 31%). A generalised linear model was used for the intention‐to‐treat analyses of each study, including all randomly assigned participants receiving at least one dose of study drug (representing 100% of those randomly assigned) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the prospectively registered protocol were reported in the published paper |
| Methods | Design: randomised, double‐blind, parallel‐group study 12‐Month study period Conducted at 94 research sites in the United States and Canada | |
| Participants | Participants: 782 people were randomly assigned to fluticasone/salmeterol combination (394) and salmeterol alone (388) Male %: flut/salm 58, salm 52 Mean age (SD), years: flut/salm 64.9 (9.0), salm 65.0 (9.1) Smoking history (mean (SD) pack‐years): flut/salm 58.5 (30.6), salm 54.4 (25.7) Mean % predicted FEV1 (SD): flut/salm 32.8 (11.0), salm 32.8 (10.1) Inclusion criteria: 40 years of age or older with a diagnosis of COPD, a cigarette smoking history of greater than or equal to 10 pack‐years, a prealbuterol FEV1/FVC of 0.70 or less, an FEV1 of 50% of predicted normal or less and a history of 1 or more exacerbations of COPD in the year before the study, which required treatment with oral corticosteroids or antibiotics, or hospitalisation | |
| Interventions | Run‐in: four‐week run‐in period during which participants received open‐label fluticasone/salmeterol 250/50 via Diskus twice daily Treatments: 1. Fluticasone/salmeterol 250/50 mcg twice daily 2. Salmterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: As‐needed albuterol was provided for use throughout the study. Concurrent inhaled long‐acting bronchodilators (beta2‐agonist and anticholinergic), ipratropium/albuterol combination products, oral beta‐agonists, inhaled corticosteroids and theophylline preparations were not allowed during the treatment period. Oral corticosteroids and antibiotics were allowed for the short‐term treatment of COPD exacerbations | |
| Outcomes | Primary: annual rate of moderate to severe exacerbations Secondary: time to first moderate to severe exacerbation, annual rate of exacerbations requiring oral corticosteroids, predose FEV1, diary records of dyspnoea, night‐time awakenings due to COPD and use of supplemental albuterol | |
| Notes | Funding: GSK (ID SCO40043) Clinicaltrials.gov ID: NCT00144911 Definition of pneumonia: no objective definition given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centre‐based randomisation schedule |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Dropout high and fairly even (30% vs 38%). Method of imputation described only for the primary outcome (‘Endpoint was defined as the last scheduled measurement of predose AM FEV1 during the 52‐week treatment period’) |
| Selective reporting (reporting bias) | Low risk | Checked GSK documents—all stated and expected outcomes are reported |
| Methods | Design: randomised, double‐blind, parallel‐group study Three‐month study period Conducted at 163 centres in 9 countries (India, Japan, Republic of Korea, Phillipines, Poland, Russian Federation, Taiwan, Ukraine and Vietnam) | |
| Participants | Participants: 1293 people were randomly assigned to budesonide/formoterol combination (636) and formoterol alone (657) Male %: bud/form 87.6, form 90.3 Mean age (range), years: bud/form 64.5 (40 to 89), form 65.6 (40 to 87) Smoking history (mean (range) pack‐years): bud/form 43.4 (10 to 160), form 44.7 (0 to 300) Mean % predicted FEV1 (range): bud/form 40.9 (12 to 79), form 40.8 (8‐84) Inclusion criteria: male and female individuals, 40 years of age or older with a diagnosis of moderate to severe COPD for at least 2 years (pre‐bronchodilator forced expiratory volume in one second (FEV1) 50% of predicted normal or less, postbronchodilator FEV1/forced vital capacity (FVC) < 70%), current or previous smoking history of 10 or more pack‐years and having at least one COPD exacerbation in 12 months | |
| Interventions | Run‐in: 1‐ to 2‐week run‐in period during which participants received open‐label formoterol 4.5 mg two inhalations twice daily and all other COPD medications were discontinued, with the exception of salbutamol 100 mg/actuation via pMDI Treatments: 1. Budesonide/formoterol combination 320/9 mcg twice daily 2. Formoterol 9 mcg twice daily Inhaler device: Turbuhaler Co‐treatment: Salbutamol 100 mg/actuation was available as reliever medication throughout the treatment period. During the randomised treatment period, participants were not permitted to take any other medication for their COPD, including beta2‐agonists, anticholinergics, leukotriene receptor antagonists, medications containing ephedrine or xanthine‐containing derivatives or inhaled corticosteroids | |
| Outcomes | Primary: change in predose FEV1 from baseline to treatment period Secondary: 1 hour postdose FEV1, predose and one hour postdose FVC, COPD symptoms, time to first COPD exacerbation, number of COPD exacerbations, health‐related quality of life on the SGRQ, morning and evening PEF, adverse events and vital signs | |
| Notes | Funding: AstraZeneca (ID: D589DC00007) Clinicaltrials.gov ID: NCT01069289 Definition of pneumonia: pneumonia, brochopneumonia and bacterial pneumonia counted as serious adverse events. Diagnosis not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned 1:1 to study treatment. No details of sequence generation methods, but presumed to adhere to usual AstraZeneca methods |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) | Low risk | From clinicaltrials.gov: masking: double‐blind (participant, caregiver, investigator, outcomes assessor) |
| Blinding of outcome assessment (detection bias) | Low risk | From clinicaltrials.gov: masking: double‐blind (participant, caregiver, investigator, outcomes assessor) |
| Incomplete outcome data (attrition bias) | Low risk | Dropout low and even between groups |
| Selective reporting (reporting bias) | Low risk | All outcomes were fully reported on clinical.trials.gov and in the published report |
| Methods | Design: multi‐centre, randomised, placebo‐controlled, double‐blind trial Three‐month treatment period Conducted at 32 centres in Germany | |
| Participants | Participants: 140 people were randomly assigned to salmeterol plus fluticasone (68) and salmeterol plus placebo (69) Male %: flut+salm 60.6, salm+placebo 71.2 Mean age (SD), years: flut+salm 61 (8), salm+placebo 63 (10) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: documented history of COPD; male and female subjects 40 to 79 years of age; smokers and ex‐smokers with a smoking history of ≥ 10 pack‐years; FEV1 40% to 80% of predicted, FEV1/FVC < 70% at visit 1 or 2; low reversibility of airway obstruction at visit 1 or 2: increase in FEV1 (normal value) < 10% at 30 minutes after inhalation of 200 mcg salbutamol; symptomatic COPD during run‐in as documented in the participant diary: on ≥ 5 days, symptom score was > 5 and/or salbutamol inhalation; ability to correctly use the Mini‐Wright Peak‐Flow‐Meter and the Diskus™ inhaler | |
| Interventions | Run‐in: 2 weeks during which all participants received salmeterol 50 mcg twice daily as bronchodilator treatment and salbutamol as rescue medication Treatments: 1. Salmeterol 50 mcg plus fluticasone 500 mcg twice daily (and placebo tablets for 2 weeks) 2. Salmeterol 50 mcg plus placebo inhaler twice daily (and placebo tablets for 2 weeks) A third group receiving oral prednisolone in combination with salmeterol was not included in this review Inhaler device: Diskus Co‐treatment: salbutamol as rescue medication | |
| Outcomes | Primary: change in FEV1 Secondary: Participants' self assessment of exercise capacity and morning serum cortisol concentrations | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described as randomised (presumed to adhere to usual GSK methodology) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Dropout relatively low and even between groups (17.6% vs 14.5%). Full analysis set (FAS) and per‐protocol analyses were reported, but only the FAS was extracted |
| Selective reporting (reporting bias) | Unclear risk | All outcomes stated were reported. Pneumonia‐related outcomes were not reported. GSK data request was not successful at the time of publication |
| Methods | Design: randomised, double‐blind, parallel‐group, comparative trial Six‐month treatment period Conducted at 55 centres in the United States | |
| Participants | Participants: 640 people were randomly assigned to fluticasone 250 (216), fluticasone 500 (218) and placebo (206) Male %: flut250 72.2, flut500 66.1, placebo 68.0 Mean age (SD), years: flut250 65.2 (8.7), flut500 63.3 (10.0), placebo 64.8 (9.5) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: Male or female individuals were eligible if they were diagnosed with COPD; were at least 40 years of age; had a current or prior history of at least 20 pack‐years of cigarette smoking; had a history of cough productive of sputum on most days for at least 3 months of the year, for at least 2 years that was not attributable to another disease process; had a baseline FEV1 < 65% of predicted normal but > 0.70 L or FEV1 ≤ 0.70 L and > 40% of predicted normal and FEV1/forced vital capacity (FVC) ratio of < 0.70; had a score of ≥ 2 on the Modified Medical Research Council (MMRC) Dyspnea Scale at screening and a score of ≥ 4 on the CBSQ at randomisation and had not received systemic corticosteroids or high‐dose inhaled corticosteroid therapy for at least 6 months before screening | |
| Interventions | Run‐in: 2‐week placebo run‐in period Treatments: 1. Fluticasone propionate 250 mcg twice daily 2. Fluticasone propionate 500 mcg twice daily 3. Placebo twice daily Inhaler device: Diskus Co‐treatment: Concurrent use of the following respiratory medications was not allowed: beta‐agonists (other than salbutamol), cromolyns, corticosteroids (oral, inhaled and intranasal), anti‐leukotrienes and ipratropium. Concurrent use of theophylline was allowed. Use of antibiotics for the treatment of up to three COPD exacerbations was allowed | |
| Outcomes | Primary: morning predose FEV1 Secondary: Chronic Bronchitis Symptoms Questionnaire (CBSQ), Transition Dyspnoea Index (TDI, exacerbations of COPD, participant‐recorded daily morning PEF rate, supplemental salbutamol use, night‐time awakenings and quality of life (CDRQ) | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised trial (GSK funded, likely to be computerised randomisation schedule) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blinded trial (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Dropout high but even across groups. The intent‐to‐treat (ITT) population consisted of all randomly assigned participants who received at least 1 dose of study medication. The ITT population was the primary population for all efficacy and safety analyses |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported in the results summary |
| Methods | Design: multi‐centre, randomised, double‐blind, double‐dummy, parallel‐group design 6 month treatment period Conducted at 135 centres in 20 countries (Australia (10), Bulgaria (5), Croatia (1), Czech Republic (8), France (14), Germany (18), Greece (4), Italy (16), Latvia (5), Lithuania (2), Netherlands (12), Philippines (3), Poland (5), Romania (3), Russian Federation (8), Slovakia (4), Slovenia (4), Sweden (4), Thailand (4) and United Kingdom (5)) | |
| Participants | Participants: 1050 people were randomly assigned to fluticasone/salmeterol combination (518) and salmeterol alone (532) Male %: flut/salm 78.4, salm 77.3 Mean age (SD), years: flut/salm 63.5 (9.4), salm 63.7 (9.0) Smoking history (mean pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: male or female, 40 to 80 years of age with an established history of GOLD stage II COPD; poor reversibility of airflow obstruction (defined as ≤ 10% increase in FEV1 as a percentage of normal predicted value); minimum score of ≥ 2 on the Modified Medical Research Council Dyspnoea Scale and a smoking history of at least 10 pack‐years. In addition, participants had to achieve a composite symptom score of ≥ 120 (out of 400 maximum score, measured using visual analogue scales) on at least 4 of the last 7 days of the run‐in period, and to have a Baseline Dyspnoea Index (BDI) score of ≤ 7 units at visit 2 | |
| Interventions | Run‐in: run‐in mentioned, unclear duration Treatments: 1. Salmeterol/fluticasone propionate 50/250 mcg twice daily 2. Salmeterol 50 mcg twice daily Inhaler device: Diskus accuhaler Co‐treatment: not reported | |
| Outcomes | Primary: FEV1, Transitional Dyspnoea Index (TDI) Secondary: change from baseline in trough FEV1, change from baseline in trough FVC and FVC/FEV1 ratio, TDI focal score, change from baseline in postdose FEV1, FVC and FVC/FEV1 ratio, change from baseline in mean morning PEF, change from baseline in St George's Respiratory Questionnaire | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described as randomised (assumed to adhere to usual GSK methodology) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described—only results summary available |
| Incomplete outcome data (attrition bias) | Low risk | Dropout low and even between groups (11.4% vs 13.9%). The ITT (intent‐to‐treat) population (all participants randomly assigned and confirmed as having received at least one dose of double‐blind study medication) was the primary population for analysis of all efficacy and health outcome variables; the safety population (identical to the ITT population) was used for analysis of all safety variables |
| Selective reporting (reporting bias) | Low risk | All stated outcomes were reported and no expected outcomes were missing |
| Methods | Design: multi‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group study Three‐month treatment period Conducted at 11 centres (4 centres in the Russian Federation, 4 centres in the United States, 2 centres in Chile and 1 centre in Estonia) | |
| Participants | Participants: 161 people were randomly assigned to fluticasone (42), placebo (42), fluticasone/salmeterol combination (39) and salmeterol (38) Male %: flut 69.0, placebo 76.2, flut/salm 82.1, salm 78.9 Mean age (SD), years: flut 64.2 (11.2), placebo 65.2 (8.6), flut/salm 63.6 (7.8), salm 64.0 (9.3) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: Males or females of non‐childbearing potential 40 years of age or older were eligible to participate if they had an established clinical history of COPD, evidence of bronchitis as a component of the COPD disease and a current or prior history of at least 10 pack‐years of cigarette smoking. Participants had a measured postalbuterol FEV1/FVC ≤ 70% at visit 1 (screening) and a measured postalbuterol FEV1 ≥ 30% and ≤ 70% of predicted normal | |
| Interventions | Run‐in: not reported Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily 3. Fluticasone/salmeterol combination 500/50 mcg twice daily 4. Salmeterol 50 mcg twice daily Inhaler device: not reported Co‐treatment: not reported | |
| Outcomes | Primary: predose resistance difference between 5 Hz and 15 Hz (R5 to R15) as measured by IOS Secondary: predose‐ and 2 hours postdose low‐frequency reactance area (AX); 2 hours postdose R5 to R15; postalbuterol computed tomography (CT) parameters of area of airway wall and area of airway lumen | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned to treatment (assumed to adhere to GSK protocol) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Withdrawal uneven between groups but all less than 20%. ITT analysis used |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in detail |
| Methods | Design: multi‐centre, randomised, double‐blind, parallel‐group, placebo‐controlled study 12‐Month treatment period Conducted at 49 centres in Italy and 7 in Poland | |
| Participants | Participants: 256 people were randomly assigned to fluticasone (131) and placebo (125) Male %: flut 83, placebo 80 Mean age (SD), years: flut 64.6 (8.7), placebo 65.7 (9.0) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: male or female individuals aged > 40 years with an established clinical history of COPD; participants who demonstrated at visit 1 a pre‐bronchodilator baseline FEV1/VC < 88% for men and < 89% for women of predicted normal values and FEV1 ≤ 70% of predicted normal value, but > 800 mL; participants who demonstrated at visit 1, poor reversibility of airflow obstruction, defined as an increase in FEV1 < 10% of the normal predicted FEV1 value (or < 200 mL from baseline), 30 minutes after inhalation of 400 mcg salbutamol via MDI; current smokers or ex‐smokers with a smoking history of at least 10 pack‐years | |
| Interventions | Run‐in: two‐week run‐in during which all inhaled corticosteroids and long‐acting beta2‐agonists were discontinued Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily Inhaler device: metered‐dose inhaler Co‐treatment: not reported | |
| Outcomes | Primary: time to first moderate or severe exacerbation Secondary: number and severity of exacerbations, withdrawals due to exacerbations, clinic FEV1, VC, FEV1/VC, daily record card symptoms, PEFR, distance walked in the six‐minute walk test (SWT), perceived breathlessness before and after SWT, quality of life (SGRQ), use of relief medication, adverse events, SAEs on therapy | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned to treatment. No details given but assumed to adhere to GSK methodology |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Dropout high at 26% and 32% for ICS and placebo, respectively. The safety population/Intent‐to‐treat (ITT) population consisted of all randomly assigned participants who took study medication (all of those randomly assigned) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the GSK summary were reported in detail |
| Methods | Design: randomised, double‐blind, parallel‐group trial Three‐year treatment period Conducted at 31 centres in the United States | |
| Participants | Participants: 186 people were randomly assigned to fluticasone/salmeterol combination (92) and salmeterol alone (94) Male %: flut/salm 59.8, salm 62.8 Mean age (SD), years: flut/salm 65.4 (8.4), salm 65.9 (9.5) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: male/female participants with an established clinical history of COPD (including a history of exacerbations), a baseline (pre‐bronchodilator) FEV1 < 70% of the predicted normal value, a baseline (pre‐bronchodilator) FEV1/FVC ratio of 70%, at least one evaluable native hip and a smoking history of ≥ 10 pack‐years | |
| Interventions | Run‐in: not reported Treatments: 1. Fluticasone propionate/salmeterol 250/50 mcg twice daily 2. Salmeterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: 'permitted COPD therapy' unclear | |
| Outcomes | Primary: change in bone mineral density at the lumbar spine Secondary: change in bone mineral density at the hip, adverse events, serious adverse events, fatal SAEs | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned to treatment (no specific information but assumed to adhere to GSK methods) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Withdrawal very high in both groups (39% and 41%) |
| Selective reporting (reporting bias) | Low risk | All outcomes described in the GSK summary were reported |
| Methods | Design: randomised, double‐blind, multi‐centre, placebo‐controlled study Six‐month treatment period Conducted at 76 investigative sites in the United States | |
| Participants | Participants: 723 people were randomly assigned to fluticasone/salmeterol combination (178), fluticasone (183), salmeterol (177) and placebo (185) Male %: flut/salm 61, flut 66, salm 58, placebo 68 Mean age (range), years: flut/salm 63 (40 to 87), flut 63 (40 to 84), salm 64 (42 to 87), placebo 65 (40 to 81) Smoking history (median (range) pack‐years): flut/salm 53 (20 to 220), flut 60 (20 to 162), salm 57 (20 to 224), placebo 56 (20 to 165) Mean % predicted FEV1 (SD): flut/salm 41 (11), flut 42 (11), salm 42 (12), placebo 42 (12) Inclusion criteria: Participants were > 40 years of age, were current or former smokers with a > 20 pack‐year history and had received a diagnosis of COPD, as defined by the American Thoracic Society. Inclusion criteria required a baseline FEV1/FVC ratio of < 70% and a baseline FEV1 of < 65% of predicted normal, but > 0.70 L (or if < 0.70 L, then > 40% of predicted normal). Participants were required to have symptoms of chronic bronchitis and moderate dyspnoea | |
| Interventions | Run‐in: two‐week, single‐blind run‐in period during which participants received placebo and albuterol and discontinued use of corticosteroids and bronchodilators with the exception of stable regimens of theophylline Treatments: 1. Fluticasone propionate 250 mcg twice daily 2. Salmeterol 50 mcg twice daily 3. Fluticasone/salmeterol 250/50 mcg twice daily 4. Placebo twice daily Inhaler device: Diskus Co‐treatment: Participants were given as‐needed albuterol and were not allowed corticosteroids or bronchodilators, with the exception of stable regimens of theophylline | |
| Outcomes | Primary: predose and 2 hours postdose FEV1 Secondary: morning PEF, dyspnoea (TDI), supplemental albuterol use, health status (CRDQ), Chronic Bronchitis Symptom Questionnaire, exacerbations, adverse events, ECG, vital signs and clinical laboratory evaluations | |
| Notes | Funding: GSK (ID: SFCA3007) Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was stratified by reversibility (no other info but GSK sponsored—likely to adhere to GSK methods) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind trial (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Dropout relatively high but even across groups. To account for participant withdrawals, endpoint was used as the primary time point and was defined as the last on‐treatment post‐baseline assessment excluding any data from the discontinuation visit |
| Selective reporting (reporting bias) | Low risk | Compared with GSK result summary and protocol—no evidence of publication bias |
| Methods | Design: double‐blind, placebo‐controlled study Three‐month study period Conducted at the London Chest Hospital | |
| Participants | Participants: 36 people were randomly assigned to fluticasone (17) and placebo (19) Male %: flut 81.3, placebo 92.9 Mean age (SD), years: flut 64.7 (6.2), placebo 64.7 (6.5) Smoking history (mean (SD) pack‐years): flut 64.9 (50.3), placebo 60.3 (46.6) Mean % predicted FEV1 (SD): flut 46.2 (13.6), placebo 45.5 (16.1) Inclusion criteria: male or female, 40 to 75 years of age, current smokers or ex‐smokers with more than 20 pack‐years of smoking, non‐atopic and with an FEV1 25% to 80% of predicted, which improved by less than 15% over baseline and 200 mL after 200 mcg inhaled salbutamol | |
| Interventions | Run‐in: After recruitment, participants had a run‐in period of 8 weeks to ensure that they were stable before the first bronchoscopy Treatments: 1. Fluticasone 500 mcg twice daily 2. Placebo twice daily Inhaler device: multi‐dose dry powder Accuhaler Co‐treatment: All reliever medications (inhaled beta2‐agonist, anticholinergics and theophylline) were continued as before | |
| Outcomes | Primary: bronchoscopy Secondary: spirometry (FEV1 and VC), biopsy processing and counts, symptom scores | |
| Notes | Funding: Glaxo‐Wellcome and Departmental Funds Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned to FP or P in a double‐blind manner using a random numbers table |
| Allocation concealment (selection bias) | Low risk | Randomly assigned in a double‐blind manner |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Seven of the randomly assigned participants did not appear in the analyses (18.9%) but were counted in the adverse event data relevant for this review |
| Selective reporting (reporting bias) | High risk | Serious adverse events not reported. No reply from study authors at time of publication |
| Methods | Design: randomised, double‐blind, parallel‐group study 10‐Month study period Conducted at 95 respiratory centres in Germany | |
| Participants | Participants: 994 people were randomly assigned to fluticasone/salmeterol combination (507) and salmeterol alone (487) Male %: flut/salm 74.0, salm 77.6 Mean age (SD), years: flut/salm 63.8 (8.3), salm 64.0 (8.2) Smoking history (mean pack‐years): flut/salm 36.8, salm 37.0 Mean % predicted FEV1 (SD): flut/salm 40.4 (8.9), salm 40.3 (8.5) Inclusion criteria: outpatients with postbronchodilator FEV1 < 50% predicted, FEV1/FVC of 70% predicted or less, age 40 years or older, smoking history of 10 or more pack‐years and a documented history of two or more moderate to severe exacerbations during the year before the study | |
| Interventions | Run‐in: four weeks Treatments: 1. Salmeterol/fluticasone 50/500 mcg twice daily 2. Salmeterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: Inhaled salbutamol was used as reliever medication, and regular treatment with short‐acting bronchodilators, antioxidants/mucolytics, short‐acting oral beta2‐agonists and theophylline was permitted | |
| Outcomes | Primary: number of exacerbations Secondary: Time to first exacerbation, pre‐bronchodilator PEF, postbronchodilator FEV1, SGRQ, symptoms and breathlessness, reliever medication use and use of other COPD medications were recorded on diary cards | |
| Notes | Funding: GSK Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Consecutive numbers assigned to participants determined the blinded treatment based on a centrally generated list with blocks of six |
| Allocation concealment (selection bias) | Low risk | Randomisation list was centrally generated |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind treatment (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Similar withdrawal rates in each group. ITT included 99.6% of the randomly assigned population (4 participants were excluded as the result of a randomisation error) |
| Selective reporting (reporting bias) | Unclear risk | Unable to locate prospective trial registration to check that all outcomes were reported. Study author contacted and forwarded request to GSK—no data were provided in time for publication |
| Methods | Design: randomised, multi‐centre, placebo‐controlled, parallel‐group trial Six‐month treatment period Conducted at 221 centres in nine countries (Chile, Estonia, Germany, Japan, Korea, Phillipines, Poland, Russian Federation and the United States) | |
| Participants | Participants: 1030 people were randomly assigned to placebo (207), fluticasone furoate 100 mcg (206), vilanterol 25 mcg (205), fluticasone furoate 50 mcg and vilanterol combination (206) and fluticasone furoate 100 mcg and vilanterol combination (206) Male %: placebo 68, flut100 64, vil 68, flut50/vil 66, flut100/vil 67 Mean age (SD), years: placebo 62.1, flut100 62.7, vil 63.4, flut50/vil 62.8, flut100/vil 62.3 Smoking history (mean (SD) pack‐years): placebo 45.6, flut100 46.2, vil 47.6, flut50/vil 44.2, flut100/vil 46.6 Mean % predicted FEV1 (SD): placebo 42.4, flut10041.5, vil 44.5, flut50/vil 42.5, flut100/vil 42.3 Inclusion criteria: over 40 years of age and have a clinical diagnosis of COPD, a smoking history of 10 pack‐years, a postbronchodilator FEV1/forced vital capacity (FVC) ratio of less than or equal to 0.70, a postbronchodilator FEV1 less than or equal to 70% predicted (NHANES III) and a score of greater than or equal to 2 on the Modified Medical Research Council Dyspnoea Scale (mMRC). A history of COPD exacerbations was not required for individuals to be eligible to enter the study. Albuterol reversibility was assessed at the screening visit, and both reversible and non‐reversible individuals were eligible to enter the study | |
| Interventions | Run‐in: 2‐week, single‐blind run‐in period during which participants received placebo once daily in the morning via a dry powder inhaler that contains two strips Treatments: 1. Fluticasone furoate 100 mcg daily 2. Vilanterol 25 mcg daily 3. Fluticasone 50 mcg/ vilanterol 25 mcg daily 4. Fluticasone 100 mcg/ vilanterol 25 mcg daily 5. Placebo Inhaler device: dry powder inhaler Co‐treatment: Albuterol, ipratropium (provided that the person was on a stable dose from the screening visit throughout the study), mucolytics, antibiotics for short‐term treatment, cough suppressants for short‐term treatment, intranasal decongestants and corticosteroids, flu and pneumonia vaccines, MAOIs, medications for other disorders as long as the dose remained constant whenever possible and their use were not expected to affect lung function | |
| Outcomes | Primary: weighted mean FEV1 (0 to 4 hours postdose) on day 168 to assess bronchodilation by FF/VI and VI (vs placebo) and FF/VI vs FF; and the change from baseline in trough (23 to 24 hours postdose) FEV1 on day 169 to assess the 24‐hour effect of VI and to determine the contribution of FF to lung function (i.e. FF/VI vs VI) Secondary: CRQ self administered standardised dyspnoea domain on day 168, various FEV1 parameters, PEF, symptom measures, adverse events, exacerbations and pneumonia events | |
| Notes | Funding: GSK (ID: HZC112206) Clinicaltrials.gov ID: NCT01053988 Definition of pneumonia: presumptive diagnosis or radiographically confirmed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated using a validated computerised system (RandAll; GlaxoSmithKline, London, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomly assigned using the Registration and Medication Ordering System (RAMOS; GlaxoSmithKline, London UK) to register and randomly assign the participant and to receive medication assignment information |
| Blinding of participants and personnel (performance bias) | Low risk | Protocol stated that the study medication was double‐blind for participant and investigator |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not stated in protocol. Some details in supplementary material about outcome assessors but unclear who was blind |
| Incomplete outcome data (attrition bias) | Low risk | Dropout was quite high but even across groups (26.7% to 33.3%). ITT population used |
| Selective reporting (reporting bias) | Low risk | All stated outcomes were available in full in the published report and on clinicaltrials.gov |
| Methods | Design: double‐blind, parallel, 4‐group, placebo‐controlled, randomised design 2.5‐Year treatment period Conducted at 2 centres in the Netherlands | |
| Participants | Participants: 55 people were randomly assigned to fluticasone (26) and placebo (29) Male %: flut 88.5, placebo 83.3 Mean age (SD), years: flut 62 (8), placebo 59 (8) Smoking history (mean (range) pack‐years): flut 44 (31 to 55), placebo 42 (34 to 54) Mean % predicted FEV1 (SD): flut 57 (9.9), placebo 54 (8.3) Inclusion criteria: 45 to 75 years of age, were current or former smokers, had smoked for 10 or more pack‐years and had lung function levels compatible with Global Initiative for Chronic Obstructive Lung Disease (GOLD) stages II and III | |
| Interventions | Run‐in: not reported Treatments: 1. Fluticasone 500 mcg twice daily 2. Placebo twice daily Inhaler device: Diskus dry powder inhaler Co‐treatment: short‐acting bronchodilators | |
| Outcomes | Primary: inflammatory cell counts in bronchial biopsies and induced sputum Secondary: postbronchodilator spirometry, hyperresponsiveness to methacholine PC20, dyspnoea score by the MRC scale, health status by the SGRQ and the Clinical COPD Questionnaire | |
| Notes | Funding: Netherlands Asthma Foundation, both centres and GSK Clinicaltrials.gov ID: NCT00158847 Definition of pneumonia: confirmed by chest x‐ray | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | At entry, an independent randomisation centre provided participant and medication numbers by using a minimisation procedure that balanced treatment groups for centre, sex, smoking status, FEV1/IVC 60% and methacholine PC20 (the provocative concentration of methacholine that causes a 20% decrease in FEV1) 2 mg/mL) |
| Allocation concealment (selection bias) | Low risk | An independent randomisation centre provided participant and medication numbers |
| Blinding of participants and personnel (performance bias) | Low risk | Study medications were individually numbered, and we used Diskus dry powder inhalers (GlaxoSmithKline, Zeist, The Netherlands) with 60 doses per inhaler; all active treatment medications and placebo were identical in appearance |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Withdrawal rates were very high compared with the numbers of events for the different outcomes. Per‐protocol analysis was used |
| Selective reporting (reporting bias) | Low risk | Data for several outcomes not available from the published report, but study authors provided data upon request |
| Methods | Design: randomised, double‐blind, parallel‐group study Six‐month treatment period | |
| Participants | Participants: 49 people were randomly assigned to budesonide (25) and placebo (24) Inclusion criteria: individuals between 40 and 65 years of age, FEV1 40% to 60% of predicted normal, FEV1/VC < 55%, bronchodilator reversibility < 15% | |
| Interventions | Run‐in: not reported Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo Inhaler device: not reported Co‐treatment: All participants received anticholinergic drug and methylxanthine or short‐acting beta2 agent | |
| Outcomes | Number and severity of exacerbations, FEV1, FVC, diary card symptoms, PEFR | |
| Notes | Abstract only Funding: not reported Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | No details provided |
| Selective reporting (reporting bias) | High risk | Only abstract available. Outcomes could not be used. Could not find contact information for study authors |
| Methods | Design: randomised, double‐blind, placebo‐controlled, parallel‐group, multi‐centre trial 6 ‐month treatment period Conducted at 65 centres | |
| Participants | Participants: 674 people were randomly assigned to fluticasone (168), placebo (181), fluticasone/salmeterol combination (165) and salmeterol alone (160) Male %: flut 61, placebo 75, flut/salm 62, salm 64 Mean age (range), years: flut 64.4 (42 to 82), placebo 64 (44 to 90), flut/salm 61.9 (40 to 86), salm 63.5 (40 to 84) Smoking history (median (range) pack‐years): flut 54 (20 to 200), placebo 60 (20 to 165), flut/salm 55 (15 to 150), salm 52.5 (20 to 193) Mean % predicted FEV1 (no SD reported): flut 41, placebo 41, flut/salm 41, salm 40 Inclusion criteria: baseline FEV1/FVC of 70% or less and baseline FEV1 of less than 65% of predicted but more than 0.70 L. Participants were required to have daily cough productive of sputum for 3 months of the year for 2 consecutive years and dyspnoea | |
| Interventions | Run‐in: 2‐week, single‐blind, run‐in period during which participants received placebo via Diskus on an as‐needed basis and discontinued use of corticosteroids and bronchodilators, with the exception of stable regimens of theophylline Treatments: 1. Fluticasone 500 mcg twice daily 2. Placebo twice daily 3. Fluticasone/Salmeterol combination 500/50 mcg twice daily 4. Salmeterol 50 mcg twice daily Inhaler device: Diskus Co‐treatment: as needed albuterol and stable regimens of theophylline | |
| Outcomes | Primary: change in predose FEV1 values and change in 2‐hour postdose FEV1 values Secondary: morning and evening PEF, supplemental albuterol use, dyspnoea as assessed by the TDI, Chronic Bronchitis Symptom Questionnaire, exacerbations defined by treatment, health status on the Chronic Respiratory Disease Questionnaire, adverse events, 24‐hour Holter monitoring and vital signs | |
| Notes | Funding: GSK (protocol number SFCA3006) Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was stratified by reversibility and investigative site to ensure balance between treatment groups at each site and in terms of the number of reversible participants (no other details, industry‐sponsored) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | 17 participants at one investigative site were not evaluable because of poor study practices, and of the remaining 674 participants, 645 had an evaluable baseline assessment. A total of 234 participants were discontinued from the study (between 28% and 40% across groups) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported in detail. Only one secondary outcome was not available |
| Methods | Design: multi‐centre, randomised, stratified (by smoking status), placebo‐controlled, double‐blind, parallel‐group study 6 month treatment period Conducted at 145 study centres in 8 countries (Czech Republic, Germany, Japan, Poland, Romania, Russian Federation, Ukraine and the United States) | |
| Participants | Participants: 1224 people were randomly assigned to fluticasone furoate 100 mcg (204), fluticasone furoate 200 mcg (203), placebo (205), fluticasone furoate 100 mcg and vilanterol combination (204), fluticasone furoate 100 mcg and vilanterol combination (205) and vilanterol (203) Male %: flut100 74, flut200 74, placebo 74, flut100/vil 71, flut200/vil 67, vil 74 Mean age (SD), years: flut100 61.8 (8.3), flut200 61.8 (9.0), placebo 61.9 (8.1), flut100/vil 61.9 (8.8), flut200/vil 61.1 (8.6), vil 61.2 (8.6) Smoking history (mean (SD) pack‐years): flut100 39.8 (21.3), flut200 43.5 (22.5), placebo 45.7 (25.8), flut100/vil 42.8 (23.9), flut200/vil 41.5 (23.4), vil 42.0 (23.3) Mean % predicted FEV1 (SD): flut100 48.4 (12.2), flut200 47.1 (12.0), placebo 48.3 (12.7), flut100/vil 48.1 (12.9), flut200/vil 47.1 (12.8), vil 48.5 (12.9) Inclusion criteria: clinical diagnosis of COPD, 40 years of age or older, smoking history of 10 or more pack‐years, postbronchodilator FEV1/ forced vital capacity (FVC) ratio of 0.70 or less, postbronchodilator FEV1 70% predicted or less (NHANES III) and score of 2 or higher on the Modified Medical Research Council Dyspnoea Scale (mMRC). No prior history of COPD exacerbations was required for individuals to be eligible to enter the study. Reversibility to albuterol was assessed at the screening visit; both reversible and non‐reversible individuals were eligible to enter the study | |
| Interventions | Run‐in: 2‐week, single‐blind run‐in period during which participants received placebo once daily in the morning via a dry powder inhaler (DPI) that contained two strips Treatments: 1. Fluticasone furoate 100 mcg daily 2. Fluticasone furoate 200 mcg daily 3. Placebo 4. Fluticasone furoate 100 mcg/vilanterol 25 mcg daily 5. Fluticasone furoate 200 mcg/vilanterol 25 mcg daily 6. Vilanterol 25 mcg Inhaler device: dry powder inhaler Co‐treatment: albuterol, ipratropium (provided that the person was on a stable dose from the screening visit throughout the study), mucolytics, antibiotics for short‐term treatment, cough suppressants for short‐term treatment, intranasal decongestants and corticosteroids, flu and pneumonia vaccines, MAOIs, medications for other disorders as long as the dose remained constant whenever possible and when their use was not expected to affect lung function | |
| Outcomes | Primary: weighted mean FEV1 (zero to four hours postdose) on day 168 to assess bronchodilation by FF/VI and VI (vs placebo) and FF/VI versus FF; change from baseline in trough (23 to 24 hours postdose) FEV1 on day 169 to assess the 24‐hour effect of VI and to determine the contribution of FF to lung function (i.e. FF/VI vs VI) Secondary: CRQ self administered standardised dyspnoea domain on day 168, various FEV parameters, PEF, symptom measures, adverse events, exacerbations and pneumonia events | |
| Notes | Funding: GSK (ID: HZC112207) Clinicaltrials.gov ID: NCT01054885 Definition of pneumonia: presumptive diagnosis or radiographically confirmed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated using a validated computerised system (RandAll; GlaxoSmithKline, London, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomly assigned using the Registration and Medication Ordering System (RAMOS; GlaxoSmithKline, London, UK) to register and randomly assign the participant and to receive medication assignment information |
| Blinding of participants and personnel (performance bias) | Low risk | Protocol stated that the study medication was double‐blind for participant and investigator |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not stated in protocol. Some details in supplementary material about outcome assessors but unclear who was blind |
| Incomplete outcome data (attrition bias) | Low risk | Dropout in all groups was less than 30% (range 20.1% to 29.4%) and an ITT analysis was used |
| Selective reporting (reporting bias) | Low risk | All stated outcomes were available in full in the published report and in supplementary tables |
| Methods | Design: randomised, placebo‐controlled, double‐blind, parallel‐group study Three‐month treatment period Conducted at a single centre in Turkey | |
| Participants | Participants: 50 people were randomly assigned to budesonide (25) and placebo (25) Male %: bud 70, placebo 80 Mean age (SD), years: bud 51.8 (9.5), placebo 54.5 (10.3) Smoking history (mean (SD) pack‐years): bud 21.7 (12.5), placebo 31.3 (19.1) Mean % predicted FEV1 (SD): bud 64.1 (6.5), placebo 59.9 (8.2) Inclusion criteria: FEV1 < 70%, FEV1 reversibility after inhalation of terbutaline from a Turbuhaler of less than 15% of pre‐bronchodilator FEV1. All participants were smokers who refused or failed a programme to quit smoking | |
| Interventions | Run‐in: not reported Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: Turbuhaler Co‐treatment: Beta2‐agonists of all kinds, theophylline and mucolytics were allowed. Inhaled corticosteroids other than study medication and oral or parenteral corticosteroids were not allowed | |
| Outcomes | Spirometry and sputum cell analysis | |
| Notes | Funding: unclear Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation sequence was computer‐generated at the Research Centre of the Faculty of Medicine |
| Allocation concealment (selection bias) | Low risk | Randomisation was masked and case numbers were allocated in consecutive order |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind. All inhalers had the same appearance and did not have drug labels |
| Blinding of outcome assessment (detection bias) | Low risk | All spirometric indices and sputum cell analyses were performed at baseline and after treatment, blind to the clinical details |
| Incomplete outcome data (attrition bias) | Unclear risk | Equal and fairly low dropout per group, values not imputed |
| Selective reporting (reporting bias) | High risk | Key expected outcomes were not reported (mortality and adverse events). Named outcomes were well reported. No reply from authors at the time of publication |
| Methods | Design: randomised, double‐blind, placebo‐controlled design Six‐month treatment period Conducted at a single centre in Turkey | |
| Participants | Participants: 26 people were randomly assigned to budesonide (13) and placebo (13) Male %: bud 84.6, placebo 53.8 Mean age (SD), years: bud 64.9 (6.1), placebo 65.9 (8.1) Smoking history (mean (SD) pack‐years): bud 45.6 (22.2), placebo 44.4 (23.0) Mean % predicted FEV1 (SD): bud 61.1 (9.7), placebo 57.3 (11.2) Inclusion criteria: (1) FEV1/FVC <70% and FEV1 50% of predicted value, (2) reversibility with inhaled beta2‐agonists (400 mg salbutamol) of less than 200 mL or less than 12% of predicted FEV1, (3) stable COPD defined as no acute exacerbation within preceding 3 months, (4) no history of systemic disease or other pulmonary disease, (5) no therapy with inhaled or systemic corticosteroids within 3 months before entry into the study and (6) no history of asthma or atopy | |
| Interventions | Run‐in: not reported Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: dry powder inhaler Co‐treatment: All participants were receiving therapy with inhaled salbutamol and ipratropium bromide. For 9 participants, sustained‐released theophyline was also given | |
| Outcomes | Primary: unclear Secondary: FVC, FEV1, diary card data, inflammatory measures | |
| Notes | Funding: unclear Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned by a computer‐generated, blinded randomisation list |
| Allocation concealment (selection bias) | Low risk | ‘Blinded’ randomisation list |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Cells were counted by our pathologist, who was also blinded. Not clear for other outcomes |
| Incomplete outcome data (attrition bias) | Unclear risk | ‘The results presented are an analysis of 22 subjects (12 budesonide‐treated subjects and 10 placebo‐treated subjects) who completed the study’. Both dropout rates were low but uneven between groups (two were excluded from the placebo group and one from the budesonide group. One extra, presumed to have been randomly assigned to placebo (assuming equal group size at randomisation) was excluded for failure to take the medication consistently) |
| Selective reporting (reporting bias) | High risk | Key expected outcomes missing (mortality, serious adverse events). No reply from study authors by time of publication |
| Methods | Design: multi‐centre, randomised, placebo‐controlled trial Six‐month treatment period Conducted in 13 European countries, New Zealand and South Africa | |
| Participants | Participants: 281 people were randomly assigned to fluticasone (142) and placebo (139) Male %: flut 70, placebo 78 Mean age (no SD reported), years: flut 62, placebo 64 Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): flut 59 (18), placebo 55 (17) Inclusion criteria: current or ex‐smokers, 50 to 75 years of age with a history of smoking equivalent to at least 10 pack‐years and chronic bronchitis (a cough with excess sputum production for at least 3 months in at least 2 consecutive years with no other pathology). Participants also had to have a history of at least one exacerbation per year for the previous 3 years that required a visit to their doctor or hospital; high expectation, according to the investigator, of experiencing an exacerbation during the 6‐month treatment period; regular productive cough; predicted FEV1 of 35% to 90%, ratio of FEV1 to forced vital capacity of 70% or less and reversibility in FEV1 of less than 15% after inhalation of 400 mcg or 800 mcg salbutamol via a metered‐dose inhaler or Diskhaler | |
| Interventions | Run‐in: 2 week run‐in period during which usual inhaled steroids were stopped and participants received salbutamol as required Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily Inhaler device: metered‐dose inhalers, with a spacer if desired Co‐treatment: Participants could take short‐acting beta2‐agonists for relief of symptoms as required throughout the study. Other COPD medications, such as anticholinergics and xanthine derivatives, could be continued throughout the study without dose changes | |
| Outcomes | Primary: COPD exacerbations Secondary: FEV1, morning PEF, FVC, 6‐minute walk test, Borg score, diary card symptom scores, daily sputum volume, total adverse events, serum cortisol concentration | |
| Notes | Funding: unclear (‘code was held by the sponsor company’s statisticians’) Definition of pneumonia:Not provided | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random numbers were computer‐generated on PACT (version 2.7) |
| Allocation concealment (selection bias) | Low risk | All investigators were given a set of four or more sealed envelopes containing assignment codes, from which they assigned treatment, starting with the lowest number |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | 'We did analysis by intention to treat of all patients who took at least one dose of study medication’ ‘Only available data was analysed’. Dropout uneven |
| Selective reporting (reporting bias) | High risk | Key expected outcomes not reported (mortality and serious adverse events). No reply from study authors by time of publication |
| Methods | Design: parallel‐group, double‐blind, placebo‐controlled, randomised, multi‐centre study 3 year treatment period Conducted at 39 study centres in nine European countries (Belgium, Denmark, Finland, Italy, the Netherlands, Norway, Spain, Sweden and the United Kingdom) | |
| Participants | Participants: 1277 people were randomly assigned to budesonide (634) and placebo (643) Male %: bud 73.5, placebo 72.2 Mean age (SD), years: bud 52.5 (7.5), placebo 52.4 (7.7) Smoking history (mean (SD) pack‐years): bud 39.4 (20.1), placebo 39.2 (20.2) Mean % predicted FEV1 (SD): bud 76.8 (12.4), placebo 76.9 (13.2) Inclusion criteria: Persons 30 to 65 years of age were eligible if they were currently smoking at least five cigarettes per day and had smoked cigarettes for at least 10 years or had a smoking history of at least 5 pack‐years. FEV1 after use of a bronchodilator had to be between 50% and 100% of predicted normal value, and ratio of pre‐bronchodilator FEV1 to slow vital capacity had to be less than 70%. Increase in FEV1 after inhalation of 1 mg of terbutaline from a dry powder inhaler had to be less than 10% of predicted normal value. Change in FEV1 between the end of the first three‐month period of the run‐in phase and the end of the second had to be less than 15% | |
| Interventions | Run‐in: three‐month smoking cessation programme. For participants who did not stop smoking, this phase was followed by a three‐month period during which compliance with inhaled medication was assessed with the use of a placebo containing dry powder inhaler with a hidden mechanical counter. Participants who continued smoking and were at least 75% compliant with the recommended treatment regimen were randomly assigned Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: 1, Pulmicort; 2, dry powder turbuhaler Co‐treatment: Use of inhaled glucocorticoids other than the study medication, beta‐blockers, cromones or long‐acting inhaled beta2‐adrenergic agonists was not allowed | |
| Outcomes | Primary: change over time in postdose FEV1 Secondary: serious adverse events, mortality, glucocorticoid‐related adverse effects, bone density, non‐serious adverse events | |
| Notes | Funding: funded by a grant from Astra Draco, Lund, Sweden Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | ‘Randomly assigned’. No specific details given but industry‐sponsored |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participant and personnel/investigator) |
| Blinding of outcome assessment (detection bias) | Low risk | Central evaluator who was unaware of the treatment received and was analysed according to a standardised computerised protocol |
| Incomplete outcome data (attrition bias) | Low risk | Data on randomly assigned participants were analysed on an intention‐to‐treat basis. Withdrawal rates under 30% and even in both groups |
| Selective reporting (reporting bias) | High risk | Several missing outcomes. Could not locate protocol to check that all prospectively registered outcomes were reported. Contacted second study author; no reply by time of publication |
| Methods | Design: parallel‐group, double‐blind, randomised, placebo‐controlled study 2‐year study period Conducted at a single centre in the Netherlands | |
| Participants | Participants: 39 people were randomly assigned to budesonide (21) and placebo (18) Male %: bud 100, placebo 100 Mean age (SD), years: bud 56 (8), placebo 54 (10) Smoking history, cigarettes/year (SD): bud 635 (530), placebo 729 (495) Mean % predicted FEV1 (SD): bud 67 (15), placebo 60 (18) Inclusion criteria: clinical diagnosis of COPD based on history (persistent dyspnoea, mainly on exertion, without sudden attacks of dyspnoea); FEV1 less than 80% of predicted value; residual volume (RV) greater than 100% of predicted value; specific compliance expressed as percentage of predicted value greater than 100% after bronchodilation; when, however, air trapping (calculated as thoracic gas volume measured by body plethysmography minus functional residual capacity measured with an indicator gas) was greater than 1.5 L Csp was allowed to be less than 100% of predicted; no signs of allergy (negative skin test results, total serum IgE < 200 IU/mL, eosinophils in peripheral blood < 250 × l03/mL); and stable phase of the disease | |
| Interventions | Run‐in: three months with no corticosteroid medication Treatments: 1. Budesonide 800 mcg twice daily (plus placebo tablet once daily) 2. Placebo twice daily (plus placebo tablet once daily) Inhaler device: metered‐dose inhaler (MDI) through a 750‐mL spacer (Nebuhaler; ASTRA, Ryswylc, The Netherlands) Co‐treatment: Throughout the study, participants were maintained on regimens of their usual bronchodilator medication, consisting of anticholinergics. beta‐agonists, theophylline or a combination of these drugs | |
| Outcomes | Primary: FEV1 Secondary: compliance, symptom scores, fasting morning plasma cortisol levels | |
| Notes | Funding: grants from the Netherlands Asthma Foundation, ASTRA BV Holland and AB DRACO Sweden Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | By computerised randomisation, stratified for smoking |
| Allocation concealment (selection bias) | Low risk | Allocated blindly |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Uneven withdrawal rates, no description of imputation to account for dropout |
| Selective reporting (reporting bias) | High risk | Key expected outcomes not reported (mortality and adverse events). No reply from study authors by time of publication |
| Methods | Design: Randomised, double‐blind, double‐dummy, parallel‐group, active‐ and placebo‐controlled, multi‐centre study 12‐Month treatment period 237 sites in the US, Europe and Mexico | |
| Participants | Participants: 1483 people were randomly assigned to budesonide/formoterol at high (494) and low dose (494), and to formoterol alone (495) Male %: bud/form high 62.3, bud/form low 62.8, form 65.3 Mean age (SD), years: bud/form high 63.2 (8.9), bud/form low 63.6 (9.2), form 62.9 (9.1) Smoking history (median pack‐years): bud/form high 40, bud/form low 40, form 40 Mean % predicted FEV1 (SD): bud/form high 38.6 (11.4), bud/form low 39.6 (10.9), form 39.3 (11.9) Inclusion criteria: current smokers or ex‐smokers aged > 40 years with clinical diagnosis of COPD and symptoms for > 2 years were eligible for this study. Participants were required to have a history of at least one COPD exacerbation treated with a course of oral corticosteroids and/or antibacterials, with 1 to 12 months before screening and documented use of an inhaled short‐acting bronchodilator as rescue medication. Prebronchodilator FEV1 of < 50% of predicted normal and pre‐bronchodilator FEV1/FVC of < 70% were required at screening. Smoking history of at least 10 pack‐years, score ≥ 2 on the Modified Medical Research Council dyspnoea scale at the time of screening and breathlessness, cough and sputum scale score ≥ 2 per day for at least half of the 2‐week run‐in period | |
| Interventions | Run‐in: 2 weeks during which previous inhaled corticosteroids were discontinued Treatments: 1. Budesonide/formoterol 320/9 mcg twice daily 2. Budesonide/formoterol 160/9 mcg twice daily 3. Formoterol 9 mcg twice daily Inhaler device: 1 and 2, pressurised metered‐dose inhaler. 3, dry powder inhaler Co‐treatment: salbutamol allowed as reliever medication | |
| Outcomes | Primary: predose FEV1 and one hour postdose FEV1 Secondary: morning and evening PEF, COPD exacerbations, quality of life (SGRQ), symptom scores, percentage of awakening‐free nights, any adverse event (AE), pneumonia‐related AEs, serious AEs, mortality, vital signs and cortisol levels | |
| Notes | Funding: AstraZeneca Clinicaltrials.gov ID: NCT00206167 Definition of pneumonia: reported by physicians based on the Medical Dictionary for Regulatory Activities (version 10.0) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Not described. Industry sponsored, presumed to follow usual AZ methods |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blind [presumed participants and personnel/investigators] |
| Blinding of outcome assessment (detection bias) | Low risk | Safety was assessed by adverse event (AE) reporting. Pneumonia events were reported by physicians based on the Medical Dictionary for Regulatory Activities (version 10.0) pneumonia‐related preferred terms (pneumonia, bronchopneumonia, lobar pneumonia or pneumonia staphylococcal). |
| Incomplete outcome data (attrition bias) | High risk | The withdrawal rates were very high compared to the number of events for the different outcomes |
| Selective reporting (reporting bias) | Low risk | All outcomes reported [checked against protocol]. Only one secondary outcome not reported. |
| Methods | Design: randomised, double‐blind, double‐dummy, placebo‐controlled phase IV trial Three‐year treatment period Conducted at 44 general practices in the Netherlands | |
| Participants | Participants: 190 people were randomly assigned to fluticasone (94) and placebo (96) Male %: flut 73, placebo 68 Mean age (SD), years: flut 58.4 (9.9), placebo 59.6 (10.1) Smoking history (mean (SD) pack‐years): flut 30.2 (18.2), placebo 26.5 (16.7) Mean % predicted FEV1 (SD): flut 63.2 (17.1), placebo 65.7 (17.7) Inclusion criteria: age 35 to 75 years; current or former smoker; chronic dyspnoea, sputum production and cough for at least three consecutive months per year during the previous two years; postbronchodilator forced expiratory volume in one second (FEV1) < 90% of predicted value and/or postbronchodilator FEV1/FVC (forced vital capacity) of predicted value < 88% for men and < 89% for women | |
| Interventions | Run‐in: Trial was preceded by the following (in chronological order): an optional smoking cessation attempt supported by the GP in trial candidates who were current smokers; a three‐month washout phase to eliminate possible carry‐over effects of a successful smoking cessation attempt or withdrawal of prior treatment with N‐acetylcysteine and/or inhaled corticosteroids; and a 14‐day pretreatment phase (30 mg oral prednisolone) to attain the highest possible baseline condition Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo twice daily Inhaler device: Diskus dry powder inhaler. Unclear from trial report whether placebo was administered to match the fluticasone inhaler or the other active treatment, which was delivered as effervescent tablets dissolved in a glass of tap Co‐treatment: not reported | |
| Outcomes | Primary: rate of exacerbation and quality of life as measured by the interviewer‐administered version of the Chronic Respiratory Questionnaire (CRQ) | |
| Notes | Funding: Dutch Council for Health Insurances, with complementary funding by the Netherlands Asthma Foundation (authors had received various GSK and other pharma research grants) Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An independent statistician generated a randomisation list based on a block size of three for treatment allocation to balance the three treatment arms by study centre. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Neither investigators nor patients were aware of the group assignment. Placebo described as ‘matching’ |
| Blinding of outcome assessment (detection bias) | Low risk | Neither investigators nor patients were aware of the group assignment [presuming the investigators were those doing the outcome assessments] |
| Incomplete outcome data (attrition bias) | High risk | Dropout was high in both groups. The primary analyses were done on an intention‐to‐treat basis. Additional per protocol analyses were done on patients with a trial medication compliance rate >80%. Unclear how data were imputed or who was included in the ITT population |
| Selective reporting (reporting bias) | High risk | All outcomes stated in the protocol were reported but some key expected outcomes were missing (serious adverse events and pneumonia). No reply from author by time of publication. |
| Methods | Design: placebo‐controlled, randomised, double‐blind, multi‐centre trial Six‐month treatment period Five centres in Denmark | |
| Participants | Participants: unclear how many people were randomly assigned. 26 were evaluable in the budesonide (14) and placebo (12) groups Male %: bud 57, placebo 50 Median age (range), years: bud 58.5 (51 to 74), placebo 62.5 (57 to 74) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): not reported Inclusion criteria: outpatients 18 to 75 years of age with stable COPD were included. FEV1, forced vital capacity (FVC) < 0.7, postbronchodilator FEV1 < 70% of predicted, FEV1 > 40% of predicted and increase in FEV1 < 15% after inhalation of 0 to 5 mg terbutaline | |
| Interventions | Run‐in: All participants received 2 weeks of treatment with oral prednisolone 37.5 mg daily. Reversible participants with 15% < AFEV1 < 30% of baseline and irreversible participants with AFEV1 < 15% were separately randomly assigned to inhaled budesonide 400 pg bid or placebo Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: Spirocort Turbuhaler Co‐treatment: not reported | |
| Outcomes | Primary: FEV1 Secondary: exacerbations, adverse events and symptom scores | |
| Notes | Funding: not reported Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Reversible participants with 15% < AFEV1 < 30% of baseline and irreversible participants with AFEV1 < 15% were separately randomly assigned |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | 26 of 37 were evaluable (12 in placebo group, 14 in active group—30% dropout overall). Number randomly assigned and number of dropouts not provided for each group. ITT not adopted |
| Selective reporting (reporting bias) | High risk | Several key expected outcomes not reported (mortality, adverse events, withdrawal per group). Study author contacted but not able to provide data |
| Methods | Design: randomised, double‐blinded, placebo‐controlled, parallel‐group, single‐centre study Two‐ to four‐year treatment period Conducted at a single centre in Denmark | |
| Participants | Participants: 254 people were randomly assigned to budesonide (127) and placebo (127) Male %: bud 62.2, placebo 54.3 Mean age (SD), years: bud 63.6 (7.5), placebo (63.6 (7.2) Smoking history (mean (SD) pack‐years): bud 56 (23), placebo 56 (24) Mean % predicted FEV1 (SD): bud 51 (11), placebo 53 (11) Inclusion criteria: Individuals 50 to 80 years of age were eligible if they were current smokers with a clinical diagnosis of COPD for not less than 2 years. All participants should have a significant smoking history of at least 10 cigarettes per day during the past 6 months and a previous history of at least 20 pack‐years. Ex‐smokers were excluded. Baseline lung function criteria were as follows: FEV1 between 35% and 70% of predicted (pre‐bronchodilator), and FEV1/forced vital capacity (FEV1/FVC) ≤ 60% | |
| Interventions | Run‐in: two‐week run‐in period on oral prednisolone Treatments: 1. Budesonide 400 mcg twice daily 2. Placebo twice daily Inhaler device: Pulmicort Turbuhaler Co‐treatment: Bronchodilators, mucolytics and short courses of oral corticosteroids (maximum 3 courses of 4 weeks' duration per year) and antibiotics were allowed during the study | |
| Outcomes | Primary: 15th percentile density (PD15) Secondary: change over time in the relative area of emphysema at a threshold of –910 Hounsfield units (RA‐910), FEV1 and diffusion capacity (DLCO) and in number of exacerbations, which was defined as a combination of 2 of the 3 following criteria: increased dyspnoea, increased sputum production and change in sputum colour | |
| Notes | Funding: AstraZeneca Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were allocated to either group in a proportion of 1:1 by block randomisation using a random sequence generated by a computer programme at AstraZeneca |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind. To maintain blinding, all Turbuhalers were of identical appearance |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | High proportion of dropouts in both groups (43% intervention and 49% placebo) |
| Selective reporting (reporting bias) | Low risk | Could not locate protocol to check that all prospectively registered outcomes were reported, but study authors provided all relevant outcomes upon request |
| Methods | Design: randomised, double‐blind, double‐dummy, parallel‐group, multi‐centre study 12‐Month treatment period Conducted at 180 study sites in the United States (106 sites), Central and South America (53 sites) and South Africa (21 sites) | |
| Participants | Participants: 1219 people were randomly assigned to high‐dose budesonide/formoterol (407), low‐dose budesonide/formoterol (408) and formoterol alone (404) Male %: bud/form high 64.4, bud/form low 64.7, form 56.8 Mean age (SD), years: bud/form high 63.8 (9.4), bud/form low 62.8 (9.2), form 62.5 (9.4) Smoking history (mean pack‐years): bud/form high 46, bud/form low 44, form 43 Mean % predicted FEV1 (SD): bud/form high 37.9 (11.8), bud/form low 37.6 (11.6), form 37.5 (12.4) Inclusion criteria: Individuals were current smokers or ex‐smokers with a smoking history of 10 pack‐years, 40 years of age, with a clinical diagnosis of COPD with symptoms for >2 years. Participants were required to have a history of 1 COPD exacerbation requiring treatment with a course of systemic corticosteroids, antibiotics or both, within one to 12 months before screening (visit 1) and documented use of an inhaled short‐acting bronchodilator as rescue medication. At screening, a pre‐bronchodilator forced expiratory volume in one second (FEV1) of 50% of predicted normal and a pre‐bronchodilator FEV1/forced vital capacity (FVC) of < 70% also were required | |
| Interventions | Run‐in: 2‐week run‐in period Treatments: 1. Budesonide/formoterol 320/9 mcg twice daily 2. Budesonide/formoterol 160/9 mcg twice daily 3. Formoterol DPI 9 mcg twice daily Inhaler device: 1 and 2, pressurised metered‐dose inhaler. 3, dry powder inhaler Co‐treatment: Rescue medication (albuterol pMDI 90 mg 2 inhalations) was provided for as needed use during screening and run‐in, and throughout the study | |
| Outcomes | Primary: COPD exacerbations Secondary: FEV1, FVC, morning and evening PEF, diary card symptoms, rescue medication use, BODE index, exercise capacity, health‐related quality of life (SGRQ), adverse events. Unclear which was primary | |
| Notes | Funding: AstraZeneca ID: D589CC00003 Clinicaltrials.gov ID: NCT00419744 Definition of pneumonia: based on clinical judgement, not on radiological or microbial assessments | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Assignments were made sequentially by interactive voice response system in a a computer generated allocation schedule produced in advance. |
| Allocation concealment (selection bias) | Low risk | Assignments were made sequentially by interactive voice response system following a computer generated allocation schedule produced in advance. |
| Blinding of participants and personnel (performance bias) | Low risk | To maintain patient and investigator blinding, all active treatments were provided in blinded treatment kits. Patients in the budesonide/formoterol pMDI groups received a placebo DPI and those in the formoterol DPI group received a placebo pMDI. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | The withdrawal rates were relatively even but high, especially compared to the low event rates for the outcomes of interest |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported in detail |
| Methods | Design: Randomised, double‐blind, placebo‐controlled, parallel‐group, multi‐centre study 12 month treatment period Conducted in 89 centres from 11 countries | |
| Participants | Participants: 812 people were randomly assigned to budesonide (198), placebo (205), budesonide/formoterol (208) and formoterol (201) Male %: bud 80, placebo 83, bud/form 76, form 76 Mean age (range), years: bud 64 (40 to 90), placebo 65 (47 to 92), bud/form 64 (41 to 82), form 63 (40 to 90) Smoking history (mean pack‐years (SD not reported)): bud 44, placebo 45, bud/form 44, form 45 Mean % predicted FEV1 (SD not reported): bud 37, placebo 36, bud/form 36, form 36 Inclusion criteria: Adults with moderate to severe asthma were included and were selected according to the following criteria: outpatients aged 0 to 40 years; COPD symptoms for 2 years; > 10 pack‐year smoking history; FEV1/FVC 70%; FEV1 50% predicted normal (stages IIB and III according to the GOLD classification); total symptom score of two per day during at least 7 days of the run‐in period; documented use of short‐acting inhaled bronchodilators for reliever medication; > 1 severe COPD exacerbation within 2 to 12 months before the first clinic visit | |
| Interventions | Run‐in: 2‐week run‐in period Treatments: 1. Budesonide 400 mcg twice daily (Pulmicort) 2. Placebo twice daily 3. Budesonide/formoterol 320/9 mcg twice daily (Symbicort) 4. Formoterol 9 mcg twice daily (Oxis) Inhaler device: as above Co‐treatment: Only study medication was allowed during the treatment period, plus terbutaline 0.5 mg when needed as reliever medication | |
| Outcomes | Primary: severe exacerbations and FEV1 Secondary: VC and PEF, health‐related quality of life, diary card data, reliever medication use, mild exacerbations, adverse events and clinical chemistry | |
| Notes | Funding: AstraZeneca Definition of pneumonia: Not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Total of 812 participants were randomly assigned (no other details, industry‐sponsored) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigator) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Withdrawal high and uneven between groups (formoterol 31.8%, placebo 43.9%, 31% ICS and 28% LABA/ICS). An intention‐to‐treat analysis was used, but imputation methods were unclear for some outcomes |
| Selective reporting (reporting bias) | Unclear risk | Could not locate trial registration to check protocol adherence—pneumonia outcomes not reported. Difficulty contacting study author to clarify |
| Methods | Design: randomised, double‐blind, double‐dummy, placebo‐controlled, parallel‐group, multi‐centre study Six‐month treatment period Conducted at 194 centres in the US, Czech Republic, the Netherlands, Poland and South Africa | |
| Participants | Participants: 1704 people were randomly assigned to six groups, but the three ICS/LABA groups were merged for analysis. Participant numbers in the four categories were as follows: budesonide (275), placebo (300), budesonide/formoterol (845) and formoterol (284) Male %: bud 67.6, placebo 69.0, bud/form 68.9, form 65.5 Mean age (SD), years: bud 63.4 (8.8), placebo 63.2 (9.6), bud/form 63.5, form 63.5 (9.5) Smoking history (mean pack‐years (SD not reported)): bud 41, placebo 40, bud/form 41, form 40 Mean % predicted FEV1 (SD): bud 39.7 (12.0), placebo 41.3 (12.1), bud/form 39.4, form 39.6 (12.8) Inclusion criteria: Current smokers or ex‐smokers older than 40 years of age with a clinical diagnosis of COPD and symptoms for longer than two years were eligible for this study. Participants were required to have a history of at least one COPD exacerbation treated with a course of oral corticosteroids and/or antibacterials with documented use of an inhaled short‐acting bronchodilator as rescue medication one to 12 months before screening. Prebronchodilator FEV1 < 50% of predicted normal and pre‐bronchodilator FEV1/FVC < 70% were required at screening. Smoking history of at least 10 pack‐years, score two or higher on the Modified Medical Research Council dyspnoea scale at the time of screening and a breathlessness, cough and sputum scale score of 2 or higher per day for at least half of the 2‐week run‐in period | |
| Interventions | Run‐in: 2‐week run‐in period Treatments: 1. Budesonide 160 mcg × 2 inhalations (320 mcg) twice daily 2. Placebo twice daily 3. Budesonide/formoterol 160/4.5 mcg × 2 inhalations (320/9 mcg) twice daily OR Budesonide/formoterol 80/4.5 mcg × 2 inhalations (160/9 mcg) twice daily OR Budesonide 160 mcg × 2 inhalations (320 mcg) twice daily plus formoterol DPI 4.5 mcg × 2 inhalations (9 mcg) twice daily 4. Formoterol DPI 4.5 mcg × 2 inhalations (9 mcg) twice daily Inhaler device: 1 and 3, pressurised metered‐dose inhaler. 4, dry powder inhaler. 2, unclear Co‐treatment: Participants were allowed the following concomitant medications during the study period: ephedrine‐free antitussives and mucolytics, nasal corticosteroids, stable‐dose non‐nebulised ipratropium bromide, oral or ophthalmic cardioselective beta‐adrenoceptor antagonists or study‐provided salbutamol as rescue medication. The following medications were allowed for exacerbations after randomisation: oral and parenteral corticosteroids, short‐term use of xanthines, increased use of inhaled beta2‐adrenoceptor agonists and ipratropium bromide, nebulised beta2‐adrenoceptor agonists and ipratropium bromide | |
| Outcomes | Primary: predose FEV1 and 1 hour postdose FEV1 Secondary: 12‐hour spirometry, predose and 1 hour postdose morning and evening PEF, dyspnoea, health‐related quality of life, COPD exacerbations, breathlessness diary and symptom scores, use of rescue medication, adverse events, serious adverse events and mortality | |
| Notes | Funding: AstraZeneca Clinicantrials.gov identifier: NCT00206154 Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Eligible participants were randomly assigned in balanced blocks according to a computer‐generated randomisation scheme at each site |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind. To maintain blinding, participants received both a pressurised metered‐dose inhaler (pMDI) and a dry powder inhaler (DPI) containing active treatment or placebo (PL), or combinations of active treatment and placebo |
| Blinding of outcome assessment (detection bias) | Unclear risk | ECG results were evaluated by a cardiologist in a blinded fashion through an independent ECG service provider. Unclear for other outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Withdrawal rates lower in combination groups (14.8% combined) than in other groups (22.9%, 25.7% and 21.5% for ICS, placebo and LABA, respectively). ITT analysis used with last observation carried forward for missing values |
| Selective reporting (reporting bias) | Low risk | Checked against protocol and contacted study authors. All stated outcomes reported in full |
| Methods | Design: randomised, placebo‐controlled, double‐blind trial 24‐Month treatment period Conducted at 10 general practices in Holland | |
| Participants | Participants: 48 people were randomly assigned to fluticasone (24) and placebo (24) Male %: flut 50, placebo 54 Mean age (SD), years: flut 46 (10), placebo 47 (11) Smoking history (mean (SD) pack‐years): flut 11.9 (9.5), placebo 5.8 (8.4) Mean % predicted FEV1 (SD): flut 95 (18), placebo 98 (17) Inclusion criteria: chronic cough and/or sputum production for at least three consecutive months and showed an annual decline in pre‐bronchodilator FEV1 of 40 to 80 mL | |
| Interventions | Run‐in: general population screened, followed by 2‐year monitoring of participants with respiratory symptoms, then randomisation Treatments: 1. Fluticasone propionate 250 mcg twice daily 2. Placebo twice daily Inhaler device: Rotadisk dry powder inhaler Co‐treatment: Apart from short‐acting ("rescue") bronchodilators in case of acute dyspnoea, participants were not allowed to use other pulmonary medication | |
| Outcomes | Primary outcome: FEV1 Secondary outcomes: PC20, exacerbations, COOP/WONCA | |
| Notes | Funding: GSK Definition of pneumonia: not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised. No details but assumed to adhere to usual GSK methods |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Dropout high but even between groups (25%). All participants with at least one follow‐up measurement for the primary outcome (postbronchodilator FEV1) were included in an intention to‐treat analysis |
| Selective reporting (reporting bias) | High risk | Key outcomes not reported (e.g. mortality). SAEs not given per arm. No reply from study authors by time of publication |
| Methods | Design: randomised, double‐blind, placebo‐controlled study Six‐month treatment period Single study centre in the Netherlands | |
| Participants | Participants: 23 people were randomly assigned to fluticasone (10) and placebo (13) Male %: flut 80, placebo 84.6 Mean age (range), years: flut 54 (42 to 65), placebo 56 (42 to 67) Smoking history (mean (range) pack‐years): flut 25 (five to 50), placebo 26 (11 to 50) Mean % predicted FEV1 (range): flut 66 (55 to 93), placebo 61 (34 to 72) Inclusion criteria: chronic productive cough, FEV1 < 70% of predicted normal value, FEV1 reversibility of < 10% predicted after 750 mg terbutaline administered by metered‐dose inhalation, negative serological examination (Phadiatop test) and negative skin prick tests for standard inhaled allergens. Individuals with an FEV1/inspiratory vital capacity (IVC) ratio of < 0.70 were also included, provided their total lung capacity (TLC) was greater than the predicted value + 1.64 SD. Participants had to be current and persistent smokers 40 to 70 years of age | |
| Interventions | Run‐in: two weeks Treatments: 1. Fluticasone propionate 500 mcg twice daily 2. Placebo Inhaler device: Diskhaler Co‐treatment: Eligible participants using anti‐inflammatory treatment including non‐steroidal anti‐inflammatory drugs were asked to refrain from oral prescriptions for at least three months and from inhaled corticosteroids, sodium cromoglycate or nedocromil sodium for at least 6 weeks before the start of the study. Long‐acting beta2‐agonists, xanthine derivatives and antihistamine drugs also had to be stopped at least 6 weeks before the start of the study | |
| Outcomes | Primary outcome unclear. Outcomes reported were use of secondary medication, compliance, FEV1, PC20, FEV1/FVC, cortisol levels and inflammatory markers | |
| Notes | Funding: GlaxoWellcome (FLIL44/FMS40060) Definition of pneumonia: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly allocated (no details, but industry‐funded) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | No dropouts in either group |
| Selective reporting (reporting bias) | Low risk | Adverse event data not adequately reported but information supplied by the study author |
| Methods | Design: double‐blind, parallel‐group, randomised clinical trial nested in a continuing epidemiological study, the Copenhagen City Heart Study (CCHS) 36‐Month treatment period Conducted at a single centre in Denmark | |
| Participants | Participants: 290 people were randomly assigned to budesonide (145) and placebo (145) Male %: bud 85, placebo 90 Mean age (SD), years: bud 59 (8.3), placebo 59.1 (9.7) Smoking history (mean (SD) pack‐years): not reported Mean % predicted FEV1 (SD): bud 86.2 (20.6), placebo 86.9 (21.1) Inclusion criteria: CCHS participant; 30 to 70 years of age; FEV1/vital capacity ratio 0.7 or less; FEV1 reversibility after inhalation of 1·0 mg terbutaline from Turbuhaler (Bricanyl, Lund, Sweden) of less than 15% of pre‐bronchodilator FEV1; FEV1 reversibility after 10 days of treatment with oral prednisolone 37.5 mg daily of less than 15% of pre‐bronchodilator FEV1; and informed consent. Pack‐years and other measures of cigarette smoking were not part of inclusion criteria | |
| Interventions | Run‐in: not described Treatments: 1. First six months: budesonide 800 mcg am and 400 mcg pm; following 30 months: 400 mcg twice daily 2. Placebo twice daily Inhaler device: Turbuhaler Co‐treatment: Continuous use of inhaled corticosteroids other than study medication was not allowed. Oral, inhaled or parenteral steroids could be used during exacerbations for up to three periods of four weeks each year. Treatment with beta2‐agonists of all kinds, theophylline, disodium cromoglycate and mucolytics was allowed but kept constant. Concomitant use of beta‐blockers during the study was not allowed | |
| Outcomes | Primary: spirometric indices (FEV1, VC, FVC) Secondary: respiratory symptoms (e.g. wheeze, wheeze without a cold, breathlessness at rest and at different grades of exertion, cough night and day, phlegm night and day, chest tightness), exacerbations, chronic mucus hypersecretion, adverse events | |
| Notes | Funding: AstraZeneca Definition of pneumonia: not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was masked and the randomisation sequence generated by computer at Astra. Study numbers were allocated in a consecutive order |
| Allocation concealment (selection bias) | Low risk | The randomisation code was held by Astra and was not available to the researchers until the study had been completed. |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind. All study inhalers (budesonide and placebo) had the same appearance. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Dropout higher in placebo group (35% vs 25% in budesonide group). ITT used. |
| Selective reporting (reporting bias) | Unclear risk | All stated outcomes reported but unable to check against trial registration. Difficulty contacting authors. |
| Methods | Design: randomised, double‐blind, placebo‐controlled design Three‐month treatment period Conducted at a single centre in Turkey | |
| Participants | Participants: 38 people randomly assigned to budesonide plus existing bronchodilator therapy (20) and placebo (18) Male %: bud 100, placebo 100 Mean age (SD), years: bud 70 (7), placebo 64 (9) Smoking history (mean (SD) pack‐years): bud 55 (31), placebo 47.5 (18) Mean % predicted FEV1 (SD): bud 51 (22), placebo 40 (14) Inclusion criteria: pre‐bronchodilator FEV1 between 30% and 80% of predicted and FEV1/FVC < 70% (stage II according to the GOLD classification), irreversible airway obstruction suggested by < 10% improvement in FEV1 after inhalation of 200 mg salbutamol, smoking history of more than 20 pack‐years and no exacerbation or respiratory tract infection in the previous four weeks | |
| Interventions | Run‐in: no information Treatments: 1. 800 mcg budesonide twice daily plus existing bronchodilator therapy 2. Placebo twice daily plus existing bronchodilator therapy Inhaler device: Miflonide inhaler, Novartis Co‐treatment: All participants were receiving combined bronchodilator therapy consisting of inhaled long‐acting beta2‐agonist (Formoterol, Foradil Aerolizer, Novartis) plus inhaled anticholinergic (ipratropium bromide, Atrovent inhaler, Boehringer Ingelheim) | |
| Outcomes | Primary: unclear which outcome was primary Secondary: St. George's Respiratory Questionnaire, FEV1, arterial blood gas analysis, serious adverse events, exacerbations | |
| Notes | Funding: unclear Definition of pneumonia: not given | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | 'Randomised'. No other details, funding unclear |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind (presumed participants and personnel/investigators) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Withdrawal rates were low (2/18 placebo, 0/20 ICS) |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported except one secondary outcome—not deemed to reflect bias. Difficulty finding correct contact details for study author |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| Treatment period less than 12 weeks | |
| Comparison not of interest (beclomethasone vs salmeterol/fluticasone combination) | |
| Asthma, not COPD | |
| Comparison not of interest (fluticasone plus oral montelukast vs salmeterol/fluticasone combination) | |
| Comparison not of interest (salmeterol/fluticasone combination vs placebo) | |
| Comparison not of interest (participants received salmeterol and salmeterol/fluticasone combination for varying amounts of time during the study) | |
| Comparison not of interest (salmeterol/fluticasone combination vs placebo) | |
| Treatment period less than 12 weeks | |
| Comparison not of interest (salmeterol/fluticasone combination vs tiotropium) | |
| Comparison not of interest (salmeterol/fluticasone combination vs tiotropium) | |
| Asthma, not COPD | |
| Comparison not of interest (triamcinolone acetonide used as inhaled steroid) | |
| ICS discontinuation study; all participants received treatment for three months before randomisation | |
| Comparison not of interest (beclomethasone used as inhaled steroid) | |
| ICS discontinuation study; all participants received treatment for three months before randomisation |
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Six‐month randomised controlled trial |
| Participants | Participants were > 40 years of age with a historical FEV1/FVC < 0.7. Six‐month history of hospitalisation attributed to AECOPD was also required |
| Interventions | Fluticasone/salmeterol combination or salmeterol alone within 14 days of an exacerbation event: < 10‐day hospitalisation for AECOPD, or AECOPD requiring treatment with OCS or OCS + antibiotics in an emergency department, or during a physician’s office visit (if the index event was office‐based) |
| Outcomes | Exacerbation rates |
| Notes | Abstract only |
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | The Study to Understand Mortality and Morbidity in COPD (SUMMIT) study protocol (NCT01313676) |
| Methods | Design: multi‐centre, placebo‐controlled, double‐blind, randomised, parallel‐group trial 15‐ to 44‐month treatment period (duration of treatment phase depended on mortality rate in the study; the study will last until 1000 deaths have been recorded) |
| Participants | Inclusion criteria: male or female, 40 to 80 years of age |
| Interventions | Run‐in: 4 to 10 days Treatments: 1. Placebo; 2. Fluticasone furoate (100 mcg once daily) 3. Vilanterol (25 mcg) 4. Fluticasone furoate/vilanterol combination (100/25 mcg once daily) Inhaler device: novel dry powder inhaler Co‐treatment: All prior use of ICS and inhaled long‐acting bronchodilators will be discontinued at entry to the run‐in period |
| Outcomes | Primary: mortality Secondary: decline in FEV1 and effect on a composite cardiovascular endpoint |
| Starting date | Recruitment commenced in March 2011 and was ongoing in March 2013 |
| Contact information | J. Vestbo, Department of Respiratory Medicine J, Odense University Hospital, Sdr Ringvej 29, 5000 Odense C, Denmark |
| Notes | None |
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Non‐fatal, serious adverse pneumonia events Show forest plot | 17 | 19504 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.78 [1.50, 2.12] |
| Analysis 1.1  Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 1 Non‐fatal, serious adverse pneumonia events. | ||||
| 1.1 Fluticasone versus placebo | 11 | 6635 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.84 [1.39, 2.44] |
| 1.2 Fluticasone/LABA versus LABA | 13 | 12869 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.75 [1.41, 2.17] |
| 2 Mortality, all‐cause Show forest plot | 22 | 20861 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.87, 1.13] |
| Analysis 1.2 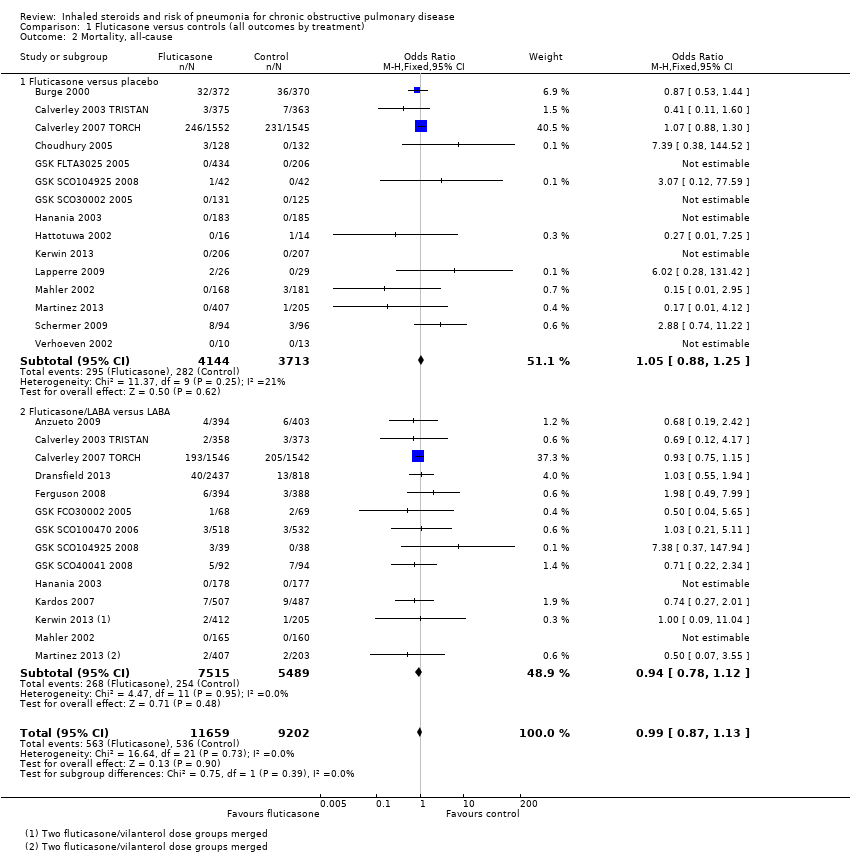 Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 2 Mortality, all‐cause. | ||||
| 2.1 Fluticasone versus placebo | 15 | 7857 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.88, 1.25] |
| 2.2 Fluticasone/LABA versus LABA | 14 | 13004 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.78, 1.12] |
| 3 Mortality, due to pneumonia Show forest plot | 18 | 19532 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.23 [0.70, 2.15] |
| Analysis 1.3  Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 3 Mortality, due to pneumonia. | ||||
| 3.1 Fluticasone versus placebo | 12 | 6665 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.20 [0.52, 2.77] |
| 3.2 Fluticasone/LABA versus LABA | 13 | 12867 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.25 [0.59, 2.65] |
| 4 Non‐fatal, serious adverse events (all) Show forest plot | 19 | 20381 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.99, 1.14] |
| Analysis 1.4 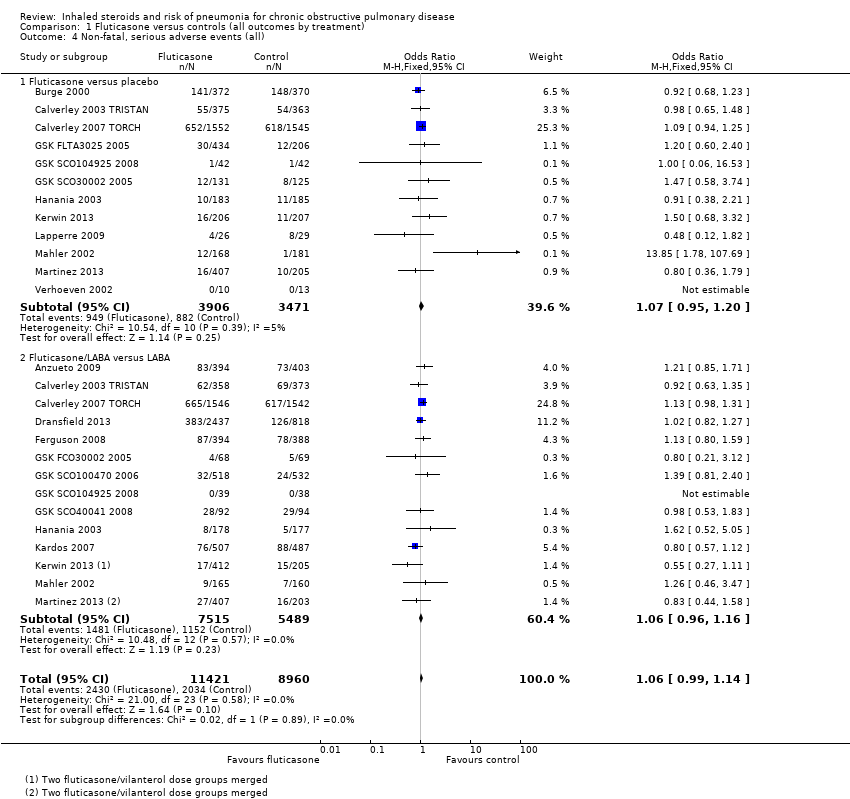 Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 4 Non‐fatal, serious adverse events (all). | ||||
| 4.1 Fluticasone versus placebo | 12 | 7377 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.95, 1.20] |
| 4.2 Fluticasone/LABA versus LABA | 14 | 13004 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.96, 1.16] |
| 5 All pneumonia events Show forest plot | 11 | 15377 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.68 [1.49, 1.90] |
| Analysis 1.5  Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 5 All pneumonia events. | ||||
| 5.1 Fluticasone versus placebo | 6 | 4971 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.33, 1.97] |
| 5.2 Fluticasone/LABA versus LABA | 9 | 10406 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.72 [1.47, 2.01] |
| 6 Withdrawals Show forest plot | 26 | 21243 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.77, 0.86] |
| Analysis 1.6 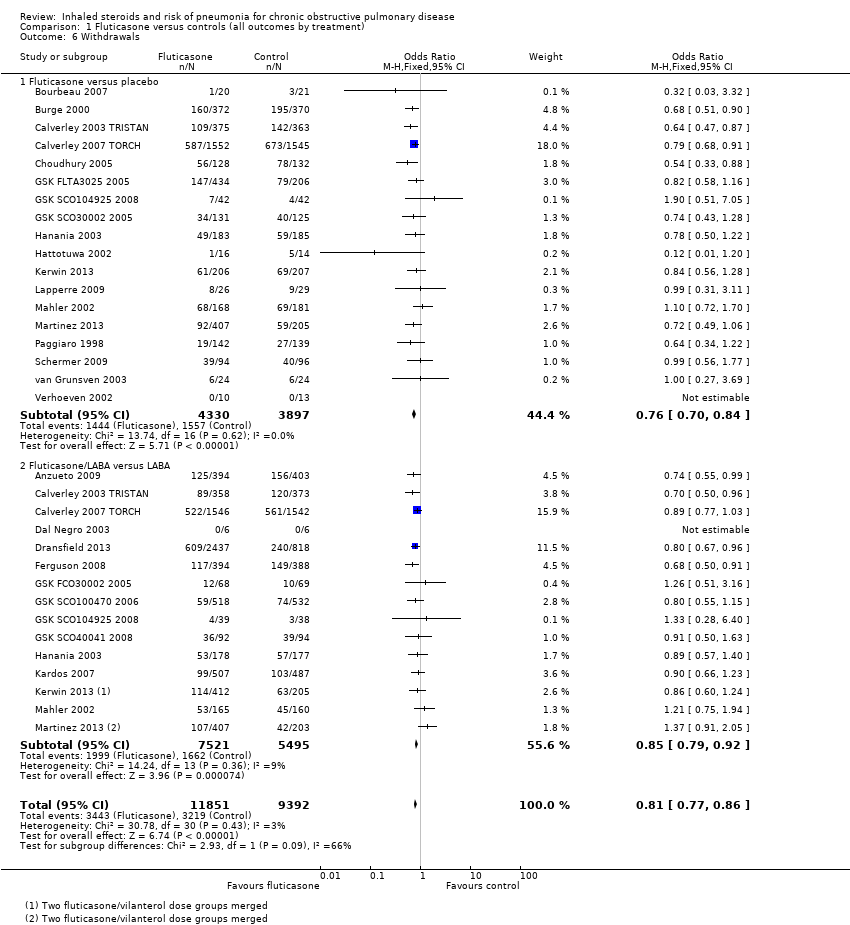 Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 6 Withdrawals. | ||||
| 6.1 Fluticasone versus placebo | 18 | 8227 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.70, 0.84] |
| 6.2 Fluticasone/LABA versus LABA | 15 | 13016 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.79, 0.92] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Dose—Non‐fatal, serious adverse pneumonia events Show forest plot | 17 | 19504 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.76 [1.48, 2.08] |
| Analysis 2.1 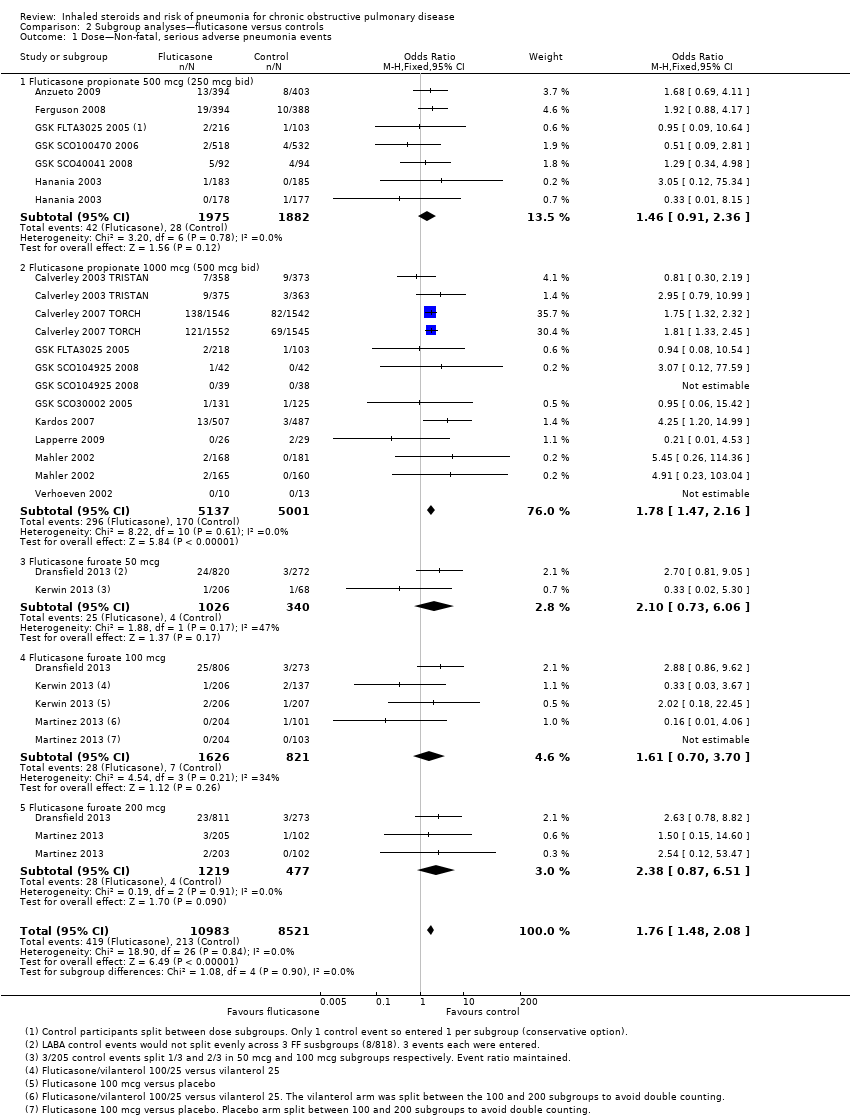 Comparison 2 Subgroup analyses—fluticasone versus controls, Outcome 1 Dose—Non‐fatal, serious adverse pneumonia events. | ||||
| 1.1 Fluticasone propionate 500 mcg (250 mcg bid) | 6 | 3857 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.91, 2.36] |
| 1.2 Fluticasone propionate 1000 mcg (500 mcg bid) | 9 | 10138 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.78 [1.47, 2.16] |
| 1.3 Fluticasone furoate 50 mcg | 2 | 1366 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.10 [0.73, 6.06] |
| 1.4 Fluticasone furoate 100 mcg | 3 | 2447 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.70, 3.70] |
| 1.5 Fluticasone furoate 200 mcg | 2 | 1696 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.38 [0.87, 6.51] |
| 2 Duration—Non‐fatal, serious adverse pneumonia events Show forest plot | 17 | 19504 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.79 [1.51, 2.12] |
| Analysis 2.2 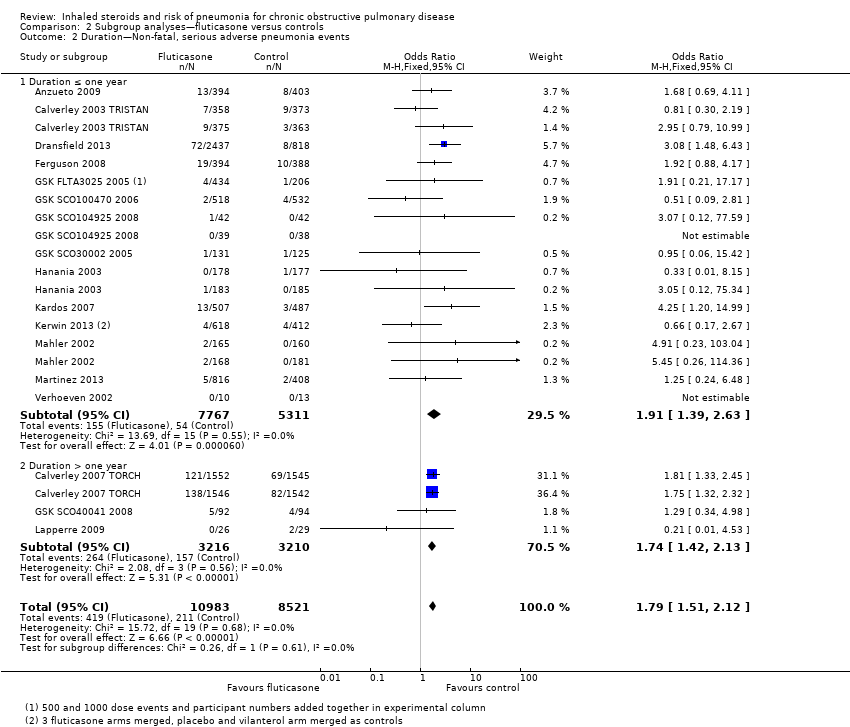 Comparison 2 Subgroup analyses—fluticasone versus controls, Outcome 2 Duration—Non‐fatal, serious adverse pneumonia events. | ||||
| 2.1 Duration ≤ one year | 14 | 13078 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.91 [1.39, 2.63] |
| 2.2 Duration > one year | 3 | 6426 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.74 [1.42, 2.13] |
| 3 % FEV1 predicted normal—Non‐fatal, serious adverse pneumonia events Show forest plot | 12 | 17211 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.82 [1.53, 2.17] |
| Analysis 2.3 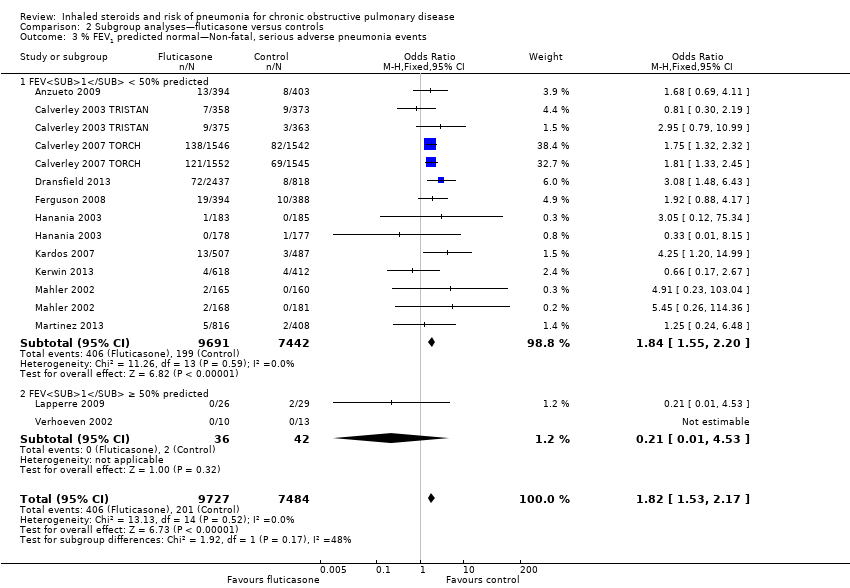 Comparison 2 Subgroup analyses—fluticasone versus controls, Outcome 3 % FEV1 predicted normal—Non‐fatal, serious adverse pneumonia events. | ||||
| 3.1 FEV1 < 50% predicted | 10 | 17133 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.84 [1.55, 2.20] |
| 3.2 FEV1 ≥ 50% predicted | 2 | 78 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.01, 4.53] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Non‐fatal, serious adverse pneumonia events Show forest plot | 7 | 6472 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.00, 2.62] |
| Analysis 3.1 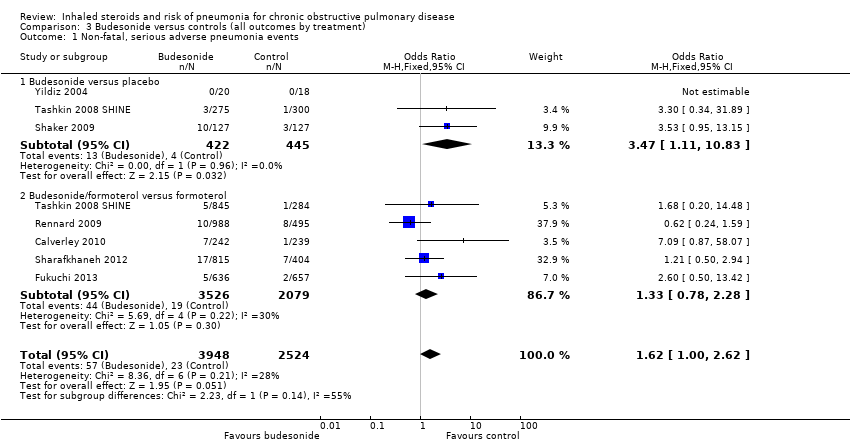 Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 1 Non‐fatal, serious adverse pneumonia events. | ||||
| 1.1 Budesonide versus placebo | 3 | 867 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.47 [1.11, 10.83] |
| 1.2 Budesonide/formoterol versus formoterol | 5 | 5605 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.78, 2.28] |
| 2 Mortality, all‐cause Show forest plot | 12 | 10009 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.65, 1.24] |
| Analysis 3.2  Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 2 Mortality, all‐cause. | ||||
| 2.1 Budesonide versus placebo | 8 | 3487 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.52, 1.37] |
| 2.2 Budesonide/formoterol versus formoterol | 7 | 6522 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.61, 1.46] |
| 3 Mortality, due to pneumonia Show forest plot | 3 | 1511 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.46 [0.07, 286.99] |
| Analysis 3.3  Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 3 Mortality, due to pneumonia. | ||||
| 3.1 Budesonide versus placebo | 2 | 292 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 Budesonide/formoterol versus formoterol | 1 | 1219 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.46 [0.07, 286.99] |
| 4 Non‐fatal, serious adverse events (all) Show forest plot | 12 | 10009 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.83, 1.22] |
| Analysis 3.4  Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 4 Non‐fatal, serious adverse events (all). | ||||
| 4.1 Budesonide versus placebo | 8 | 3487 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.69, 1.50] |
| 4.2 Budesonide/formoterol versus formoterol | 7 | 6522 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.78, 1.11] |
| 5 All pneumonia events Show forest plot | 6 | 7011 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.83, 1.51] |
| Analysis 3.5  Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 5 All pneumonia events. | ||||
| 5.1 Budesonide versus placebo | 3 | 1378 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.50, 1.50] |
| 5.2 Budesonide/formoterol versus formoterol | 5 | 5633 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.87, 1.77] |
| 6 Withdrawals Show forest plot | 15 | 10150 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.71, 0.85] |
| Analysis 3.6 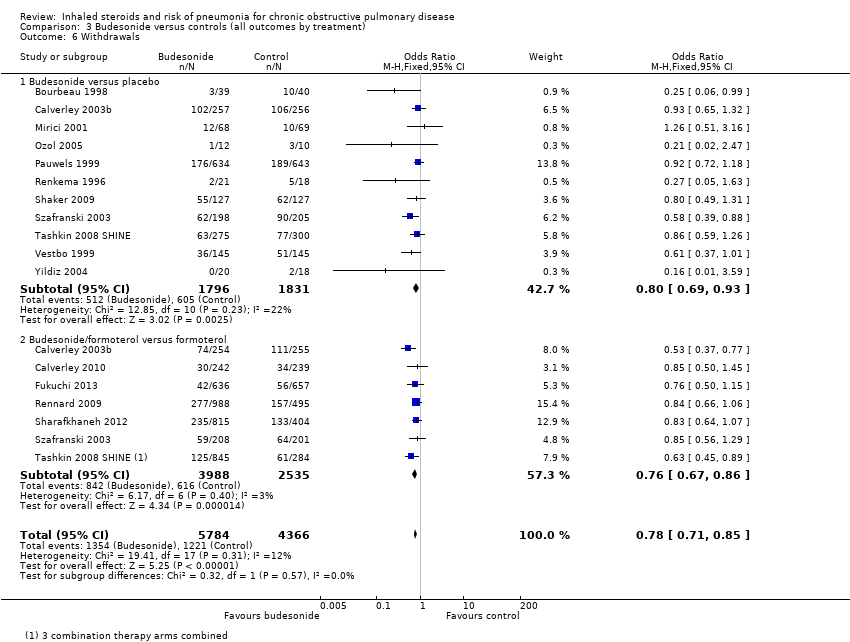 Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 6 Withdrawals. | ||||
| 6.1 Budesonide versus placebo | 11 | 3627 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.69, 0.93] |
| 6.2 Budesonide/formoterol versus formoterol | 7 | 6523 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.67, 0.86] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Dose ‐ Non‐fatal, serious adverse pneumonia events Show forest plot | 7 | 6472 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.54 [0.96, 2.48] |
| Analysis 4.1 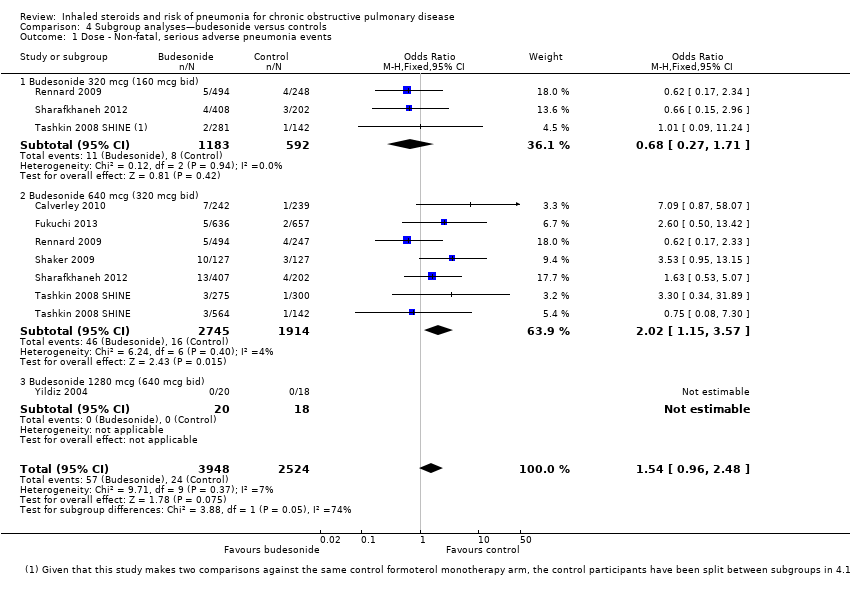 Comparison 4 Subgroup analyses—budesonide versus controls, Outcome 1 Dose ‐ Non‐fatal, serious adverse pneumonia events. | ||||
| 1.1 Budesonide 320 mcg (160 mcg bid) | 3 | 1775 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.27, 1.71] |
| 1.2 Budesonide 640 mcg (320 mcg bid) | 6 | 4659 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.02 [1.15, 3.57] |
| 1.3 Budesonide 1280 mcg (640 mcg bid) | 1 | 38 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Duration ‐ Non‐fatal, serious adverse pneumonia events Show forest plot | 7 | 6471 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.00, 2.62] |
| Analysis 4.2  Comparison 4 Subgroup analyses—budesonide versus controls, Outcome 2 Duration ‐ Non‐fatal, serious adverse pneumonia events. | ||||
| 2.1 Duration ≤ one year | 6 | 6217 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.41 [0.83, 2.37] |
| 2.2 Duration > one year | 1 | 254 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.53 [0.95, 13.15] |
| 3 % FEV1 predicted normal ‐ Non‐fatal, serious adverse pneumonia events Show forest plot | 7 | 6471 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.60 [0.99, 2.59] |
| Analysis 4.3  Comparison 4 Subgroup analyses—budesonide versus controls, Outcome 3 % FEV1 predicted normal ‐ Non‐fatal, serious adverse pneumonia events. | ||||
| 3.1 FEV1 < 50% predicted | 6 | 6217 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.82, 2.34] |
| 3.2 FEV1 ≥ 50% predicted | 1 | 254 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.53 [0.95, 13.15] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Non‐fatal serious adverse pneumonia events Show forest plot | 16 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 5.1 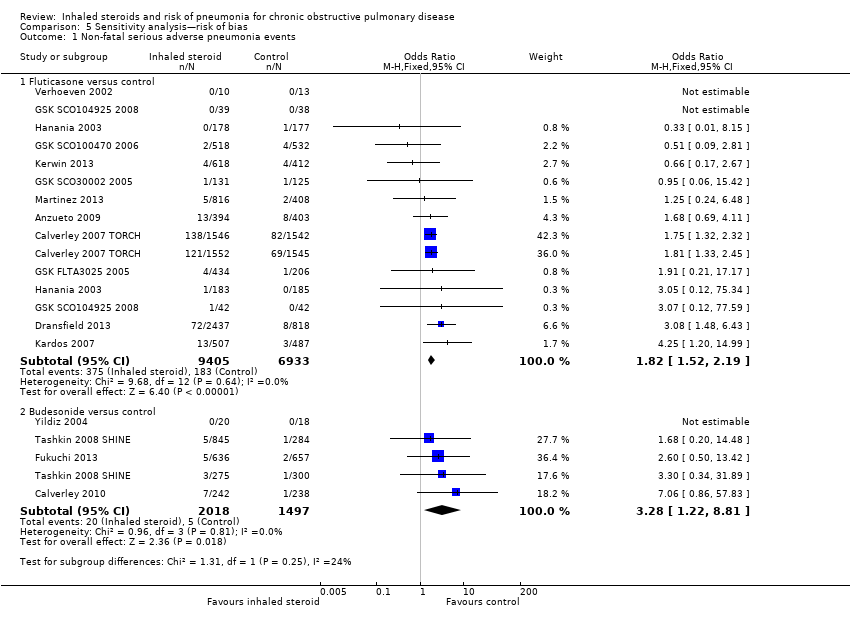 Comparison 5 Sensitivity analysis—risk of bias, Outcome 1 Non‐fatal serious adverse pneumonia events. | ||||
| 1.1 Fluticasone versus control | 12 | 16338 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.82 [1.52, 2.19] |
| 1.2 Budesonide versus control | 4 | 3515 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.28 [1.22, 8.81] |
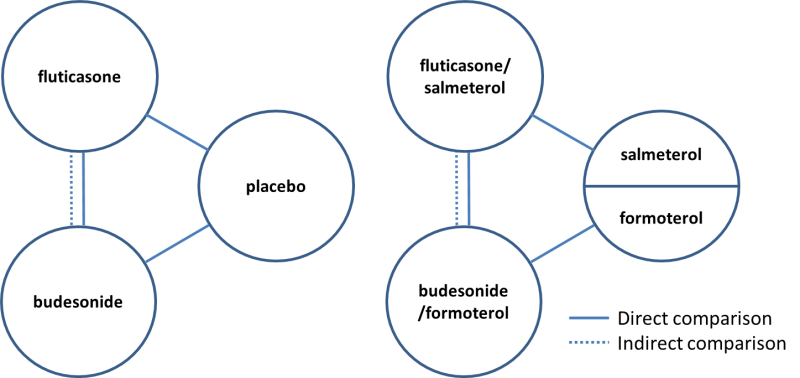
Direct and indirect comparisons of fluticasone and budesonide covered in the review.


Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
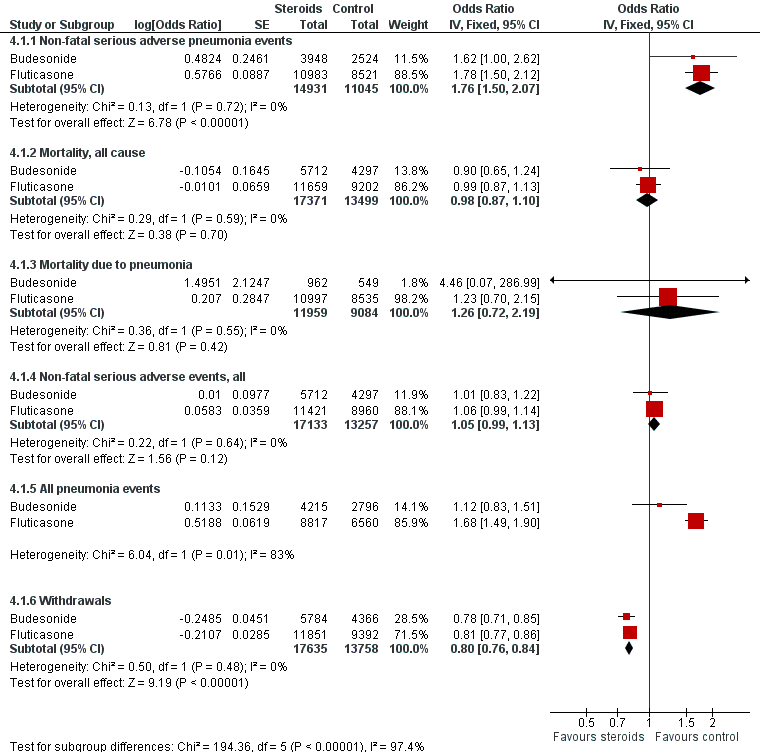
Summary of pooled effects of trials comparing ICS versus placebo and combination versus LABA.
Non‐fatal serious adverse pneumonia events were those requiring hospital admission. Data for all pneumonia events were not pooled because of heterogeneity.

Indirect comparisons of fluticasone and budesonide monotherapy.
Non‐fatal serious adverse pneumonia events were defined as those requiring hospital admission.

Indirect comparisons of fluticasone and budesonide monotherapy—Sensitivity analysis removing (Calverley 2007 TORCH).
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 1 Non‐fatal, serious adverse pneumonia events.
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 2 Mortality, all‐cause.
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 3 Mortality, due to pneumonia.
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 4 Non‐fatal, serious adverse events (all).
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 5 All pneumonia events.
Comparison 1 Fluticasone versus controls (all outcomes by treatment), Outcome 6 Withdrawals.
Comparison 2 Subgroup analyses—fluticasone versus controls, Outcome 1 Dose—Non‐fatal, serious adverse pneumonia events.
Comparison 2 Subgroup analyses—fluticasone versus controls, Outcome 2 Duration—Non‐fatal, serious adverse pneumonia events.
Comparison 2 Subgroup analyses—fluticasone versus controls, Outcome 3 % FEV1 predicted normal—Non‐fatal, serious adverse pneumonia events.
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 1 Non‐fatal, serious adverse pneumonia events.
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 2 Mortality, all‐cause.
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 3 Mortality, due to pneumonia.
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 4 Non‐fatal, serious adverse events (all).
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 5 All pneumonia events.
Comparison 3 Budesonide versus controls (all outcomes by treatment), Outcome 6 Withdrawals.
Comparison 4 Subgroup analyses—budesonide versus controls, Outcome 1 Dose ‐ Non‐fatal, serious adverse pneumonia events.
Comparison 4 Subgroup analyses—budesonide versus controls, Outcome 2 Duration ‐ Non‐fatal, serious adverse pneumonia events.
Comparison 4 Subgroup analyses—budesonide versus controls, Outcome 3 % FEV1 predicted normal ‐ Non‐fatal, serious adverse pneumonia events.
Comparison 5 Sensitivity analysis—risk of bias, Outcome 1 Non‐fatal serious adverse pneumonia events.
| Fluticasone for chronic obstructive pulmonary disease | |||||
| Patient or population: patients with chronic obstructive pulmonary disease Comparison: placebo or LABA monotherapy (dependent upon whether fluticasone was given with LABA in the intervention group) Setting: community | |||||
| Outcomes Follow‐ups presented as weighted means | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | |
| Assumed risk | Corresponding risk | ||||
| Control | Fluticasone | ||||
| Non‐fatal, serious adverse pneumonia events (requiring hospital admission) | 25 per 1000 | 43 per 1000 | OR 1.78 | 19,504 | ⊕⊕⊕⊕ |
| Mortality, all‐cause | 58 per 1000 | 58 per 1000 | OR 0.99 | 20,861 | ⊕⊕⊕⊕ |
| Mortality, due to pneumonia | 2 per 1000 | 3 per 1000 | OR 1.23 | 19,532 | ⊕⊕⊕⊝ |
| Non‐fatal, serious adverse events (all) | 227 per 1000 | 237 per 1000 | OR 1.06 | 20,381 | ⊕⊕⊕⊕ |
| All pneumonia events | 72 per 1000 | 116 per 1000 | OR 1.68 | 15,377 | ⊕⊕⊕⊝ |
| Withdrawals | 343 per 1000 | 297 per 1000 | OR 0.81 | 21,243 | ⊕⊕⊕⊕ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Unless otherwise stated, subgroup differences between monotherapy studies (fluticasone versus placebo) and combination therapy studies (fluticasone/LABA versus LABA) were not significant. | |||||
| GRADE Working Group grades of evidence. | |||||
| 1Wide confidence intervals include significant benefit and harm, based on very few events (‐1 for imprecision). | |||||
| Budesonide for chronic obstructive pulmonary disease | |||||
| Patient or population: patients with chronic obstructive pulmonary disease Comparison: placebo or LABA monotherapy (dependent upon whether fluticasone was given with LABA in the intervention group) Setting: community | |||||
| Outcomes Follow‐ups presented as weighted means | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | |
| Assumed risk | Corresponding risk | ||||
| Control | Budesonide | ||||
| Non‐fatal, serious adverse pneumonia events (requiring hospital admission) | 9 per 1000 | 15 per 1000 | OR 1.62 | 6472 | ⊕⊕⊕⊝ |
| Mortality, all‐cause | 17 per 1000 | 16 per 1000 | OR 0.90 | 10,009 | ⊕⊕⊕⊝ |
| Mortality, due to pneumonia | 0 per 1000 | 0 per 1000 | OR 4.46 | 1511 | ⊕⊝⊝⊝ |
| Non‐fatal, serious adverse events (all) | 145 per 1000 | 146 per 1000 | OR 1.01 | 10,009 | ⊕⊕⊕⊝ |
| All pneumonia events | 28 per 1000 | 31 per 1000 | OR 1.12 | 7011 | ⊕⊕⊕⊝ |
| Withdrawals | 280 per 1000 | 232 per 1000 | OR 0.78 | 10150 | ⊕⊕⊕⊕ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Unless otherwise stated, subgroup differences between monotherapy studies (budesonide versus placebo) and combination therapy studies (budesonide/LABA versus LABA) were not significant. | |||||
| GRADE Working Group grades of evidence. | |||||
| 1Confidence intervals are quite wide but are not considered serious enough to downgrade. | |||||
| Study ID | Duration (m) | N Rand | Funder | ICS dose (mcg) | % Male | Mean age | Pack‐years | % pred FEV1 |
| Fluticasone versus placebo (n = 18) | ||||||||
| 3 | 41 | GSK | 1000 | 78 | 65 | 53 | 57 | |
| 36 | 740 | GSK | 1000 | 75 | 64 | 44 | 50 | |
| 12 | 763 | GSK | 1000 | 73 | 63 | 43 | 45 | |
| 36 | 3097 | GSK | 1000 | 76 | 65 | 49 | 44 | |
| 12 | 260 | Indep. | 1000 | 52 | 67 | 39 | 54 | |
| 6 | 640 | GSK | 500, 1000 | 69 | 64 | ‐ | ‐ | |
| 3 | 84 | GSK | 1000 | 77 | 64 | ‐ | ‐ | |
| 12 | 256 | GSK | 1000 | 82 | 65 | ‐ | ‐ | |
| 6 | 368 | GSK | 500 | 63 | 64 | 57 | 42 | |
| 3 | 36 | GSK | 1000 | 87 | 65 | 63 | 46 | |
| Kerwin 2013a,b | 6 | 413 | GSK | 100 | 66 | 62 | 46 | 42 |
| 30 | 55 | GSK | 1000 | 86 | 60 | 43 | 55 | |
| 6 | 349 | GSK | 1000 | 66 | 65 | 55 | 41 | |
| 6 | 612 | GSK | 100, 200 | 72 | 62 | 43 | 48 | |
| 6 | 281 | ‐ | 1000 | 74 | 63 | ‐ | 57 | |
| 36 | 190 | Indep. | 1000 | 71 | 59 | 28 | 64 | |
| 24 | 48 | GSK | 500 | 52 | 47 | 9 | 97 | |
| 6 | 23 | GSK | 1000 | 82 | 55 | 26 | 63 | |
| WM 22 m | 459 | ‐ | ‐ | 72 | 62 | 43 | 54 | |
| Fluticasone/LABA combination versus LABA monotherapy (n = 15) | ||||||||
| 12 | 797 | GSK | 500 | 54 | 65 | 57 | 34 | |
| 12 | 731 | GSK | 1000 | 73 | 63 | 43 | 45 | |
| 36 | 3088 | GSK | 1000 | 76 | 65 | 49 | 44 | |
| 12 | 12 | ‐ | 500 | 92 | ‐ | 42 | 50 | |
| 12 | 3255 | GSK | 50, 100, 200 | 57 | 64 | ‐ | 45 | |
| 12 | 782 | GSK | 500 | 55 | 65 | 56 | 33 | |
| 3 | 140 | GSK | 1000 | 66 | 62 | ‐ | ‐ | |
| 6 | 1050 | GSK | 500 | 78 | 64 | ‐ | ‐ | |
| 3 | 77 | GSK | 1000 | 77 | 64 | ‐ | ‐ | |
| 36 | 186 | GSK | 500 | 61 | 66 | ‐ | ‐ | |
| 6 | 355 | GSK | 500 | 63 | 64 | 57 | 42 | |
| 10 | 994 | GSK | 1000 | 76 | 64 | 37 | 40 | |
| Kerwin 2013a,b | 6 | 617 | GSK | 100 | 67 | 63 | 46 | 43 |
| 6 | 325 | GSK | 1000 | 66 | 65 | 55 | 41 | |
| 6 | 610 | GSK | 100, 200 | 72 | 62 | 43 | 48 | |
| WM 16 m | 867 | ‐ | ‐ | 69 | 64 | 53 | 42 | |
| aMulti‐arm studies making both comparisons of interest (ICS vs placebo and ICS/LABA vs LABA). bStudies using vilanterol as the LABA combination and monotherapy comparator, with fluticasone furoate. Dose is given as the total received per day (i.e. 500 signifies 250 morning and evening). WM = weighted mean. | ||||||||
| Study ID | Duration (m) | N Rand | Funder | ICS dose (mcg) | % Male | Mean age | Pack‐years | % pred FEV1 |
| Budesonide versus placebo (n = 13) | ||||||||
| 6 | 79 | AZ | 640 | 79 | 66 | 51 | 37 | |
| 12 | 513 | GSK | 640 | 76 | 64 | 35 | 36 | |
| 6 | 49 | NR | 640 | NR | NR | NR | NR | |
| 3 | 50 | NR | 640 | 75 | 53 | 27 | 62 | |
| 6 | 26 | NR | 640 | 69 | 65 | 45 | 59 | |
| 36 | 1277 | AZ | 640 | 73 | 52 | 39 | 77 | |
| 24 | 39 | AZ | 1280 | 100 | 55 | NR | 64 | |
| 6 | 26 | NR | 640 | 54 | 61 | NR | NR | |
| 36 | 254 | AZ | 640 | 58 | 64 | 56 | 52 | |
| 12 | 403 | AZ | 640 | 79 | 64 | 45 | 36 | |
| 6 | 575 | AZ | 640 | 68 | 63 | 41 | 40 | |
| 36 | 290 | AZ | 640 | 88 | 59 | NR | 87 | |
| 3 | 38 | ? | 1280 | 100 | 67 | 51 | 46 | |
| WM 23 m | 278 | ‐ | ‐ | 77 | 61 | 43 | 54 | |
| Budesonide/LABA combination versus LABA monotherapy (n = 7) | ||||||||
| 12 | 509 | GSK | 640 | 76 | 64 | 35 | 36 | |
| 11 | 481 | Chiesi | 640 | 81 | 64 | 39 | 42 | |
| 3 | 1293 | AZ | 640 | 89 | 65 | 44 | 41 | |
| 12 | 1483 | AZ | 320, 640 | 63 | 63 | NR | 39 | |
| 12 | 1219 | AZ | 320, 640 | 62 | 63 | 44 | 38 | |
| 12 | 409 | AZ | 640 | 79 | 64 | 45 | 36 | |
| 6 | 1129 | AZ | 320, 640 | 68 | 63 | 41 | 40 | |
| WM 9 m | 932 | ‐ | ‐ | 75 | 64 | 41 | 39 | |
| aMulti‐arm studies making both comparisons of interest (ICS vs placebo and ICS/LABA vs LABA). Dose is given as the total received per day (i.e. 640 signifies 320 morning and evening). WM = weighted mean. | ||||||||
| Drug | Daily dose (mcg) | BDP equivalent (mcg) |
| Budesonide | 320 | 320 |
| 640 | 640 | |
| 1280 | 1280 | |
| Fluticasone | 500 (propionate) | 1000 |
| 1000 (propionate) | 2000 | |
| 50 (furoate) | 500 | |
| 100 (furoate) | 1000 | |
| 200 (furoate) | 2000 |
| Monotherapy comparison—Placebo control events | Combination comparison—LABA control events | |||
| Fluticasone | Budesonide | Fluticasone | Budesonide | |
| Pneumonia‐related serious adverse events | 2.5%, 77/310 | 0.9%, 4/445 | 2.5%, 134/5420 | 0.9%, 19/2079 |
| 0.5% without TORCH | 0.7% without TORCH | |||
| All‐cause mortality | 7.6%, 282/3713 | 2.1%, 37/1763 | 5.1%, 254/5489 | 1.5%, 37/2534 |
| 2.4% without TORCH | 1.2% without TORCH | |||
| All‐cause serious adverse events | 25%, 882/3471 | 15%, 268/1763 | 21%, 1152/5489 | 14%, 356/2534 |
| 14% without TORCH | 14% without TORCH | |||
| For the fluticasone control groups with and without the large 3‐year TORCH study. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Non‐fatal, serious adverse pneumonia events Show forest plot | 17 | 19504 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.78 [1.50, 2.12] |
| 1.1 Fluticasone versus placebo | 11 | 6635 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.84 [1.39, 2.44] |
| 1.2 Fluticasone/LABA versus LABA | 13 | 12869 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.75 [1.41, 2.17] |
| 2 Mortality, all‐cause Show forest plot | 22 | 20861 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.87, 1.13] |
| 2.1 Fluticasone versus placebo | 15 | 7857 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.88, 1.25] |
| 2.2 Fluticasone/LABA versus LABA | 14 | 13004 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.78, 1.12] |
| 3 Mortality, due to pneumonia Show forest plot | 18 | 19532 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.23 [0.70, 2.15] |
| 3.1 Fluticasone versus placebo | 12 | 6665 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.20 [0.52, 2.77] |
| 3.2 Fluticasone/LABA versus LABA | 13 | 12867 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.25 [0.59, 2.65] |
| 4 Non‐fatal, serious adverse events (all) Show forest plot | 19 | 20381 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.99, 1.14] |
| 4.1 Fluticasone versus placebo | 12 | 7377 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.95, 1.20] |
| 4.2 Fluticasone/LABA versus LABA | 14 | 13004 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.96, 1.16] |
| 5 All pneumonia events Show forest plot | 11 | 15377 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.68 [1.49, 1.90] |
| 5.1 Fluticasone versus placebo | 6 | 4971 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.33, 1.97] |
| 5.2 Fluticasone/LABA versus LABA | 9 | 10406 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.72 [1.47, 2.01] |
| 6 Withdrawals Show forest plot | 26 | 21243 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.77, 0.86] |
| 6.1 Fluticasone versus placebo | 18 | 8227 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.70, 0.84] |
| 6.2 Fluticasone/LABA versus LABA | 15 | 13016 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.79, 0.92] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Dose—Non‐fatal, serious adverse pneumonia events Show forest plot | 17 | 19504 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.76 [1.48, 2.08] |
| 1.1 Fluticasone propionate 500 mcg (250 mcg bid) | 6 | 3857 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.91, 2.36] |
| 1.2 Fluticasone propionate 1000 mcg (500 mcg bid) | 9 | 10138 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.78 [1.47, 2.16] |
| 1.3 Fluticasone furoate 50 mcg | 2 | 1366 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.10 [0.73, 6.06] |
| 1.4 Fluticasone furoate 100 mcg | 3 | 2447 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.70, 3.70] |
| 1.5 Fluticasone furoate 200 mcg | 2 | 1696 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.38 [0.87, 6.51] |
| 2 Duration—Non‐fatal, serious adverse pneumonia events Show forest plot | 17 | 19504 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.79 [1.51, 2.12] |
| 2.1 Duration ≤ one year | 14 | 13078 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.91 [1.39, 2.63] |
| 2.2 Duration > one year | 3 | 6426 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.74 [1.42, 2.13] |
| 3 % FEV1 predicted normal—Non‐fatal, serious adverse pneumonia events Show forest plot | 12 | 17211 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.82 [1.53, 2.17] |
| 3.1 FEV1 < 50% predicted | 10 | 17133 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.84 [1.55, 2.20] |
| 3.2 FEV1 ≥ 50% predicted | 2 | 78 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.01, 4.53] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Non‐fatal, serious adverse pneumonia events Show forest plot | 7 | 6472 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.00, 2.62] |
| 1.1 Budesonide versus placebo | 3 | 867 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.47 [1.11, 10.83] |
| 1.2 Budesonide/formoterol versus formoterol | 5 | 5605 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.78, 2.28] |
| 2 Mortality, all‐cause Show forest plot | 12 | 10009 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.65, 1.24] |
| 2.1 Budesonide versus placebo | 8 | 3487 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.52, 1.37] |
| 2.2 Budesonide/formoterol versus formoterol | 7 | 6522 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.61, 1.46] |
| 3 Mortality, due to pneumonia Show forest plot | 3 | 1511 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.46 [0.07, 286.99] |
| 3.1 Budesonide versus placebo | 2 | 292 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 Budesonide/formoterol versus formoterol | 1 | 1219 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.46 [0.07, 286.99] |
| 4 Non‐fatal, serious adverse events (all) Show forest plot | 12 | 10009 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.83, 1.22] |
| 4.1 Budesonide versus placebo | 8 | 3487 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.69, 1.50] |
| 4.2 Budesonide/formoterol versus formoterol | 7 | 6522 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.78, 1.11] |
| 5 All pneumonia events Show forest plot | 6 | 7011 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.83, 1.51] |
| 5.1 Budesonide versus placebo | 3 | 1378 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.50, 1.50] |
| 5.2 Budesonide/formoterol versus formoterol | 5 | 5633 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.87, 1.77] |
| 6 Withdrawals Show forest plot | 15 | 10150 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.71, 0.85] |
| 6.1 Budesonide versus placebo | 11 | 3627 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.69, 0.93] |
| 6.2 Budesonide/formoterol versus formoterol | 7 | 6523 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.67, 0.86] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Dose ‐ Non‐fatal, serious adverse pneumonia events Show forest plot | 7 | 6472 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.54 [0.96, 2.48] |
| 1.1 Budesonide 320 mcg (160 mcg bid) | 3 | 1775 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.27, 1.71] |
| 1.2 Budesonide 640 mcg (320 mcg bid) | 6 | 4659 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.02 [1.15, 3.57] |
| 1.3 Budesonide 1280 mcg (640 mcg bid) | 1 | 38 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Duration ‐ Non‐fatal, serious adverse pneumonia events Show forest plot | 7 | 6471 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.00, 2.62] |
| 2.1 Duration ≤ one year | 6 | 6217 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.41 [0.83, 2.37] |
| 2.2 Duration > one year | 1 | 254 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.53 [0.95, 13.15] |
| 3 % FEV1 predicted normal ‐ Non‐fatal, serious adverse pneumonia events Show forest plot | 7 | 6471 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.60 [0.99, 2.59] |
| 3.1 FEV1 < 50% predicted | 6 | 6217 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.82, 2.34] |
| 3.2 FEV1 ≥ 50% predicted | 1 | 254 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.53 [0.95, 13.15] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Non‐fatal serious adverse pneumonia events Show forest plot | 16 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Fluticasone versus control | 12 | 16338 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.82 [1.52, 2.19] |
| 1.2 Budesonide versus control | 4 | 3515 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.28 [1.22, 8.81] |